


Thalassemia inheritance pattern explained helps readers understand how this inherited blood disorder is passed through autosomal recessive genes, what carrier status means, and why genetic testing and family planning are essential for early diagnosis and informed healthcare decisions.
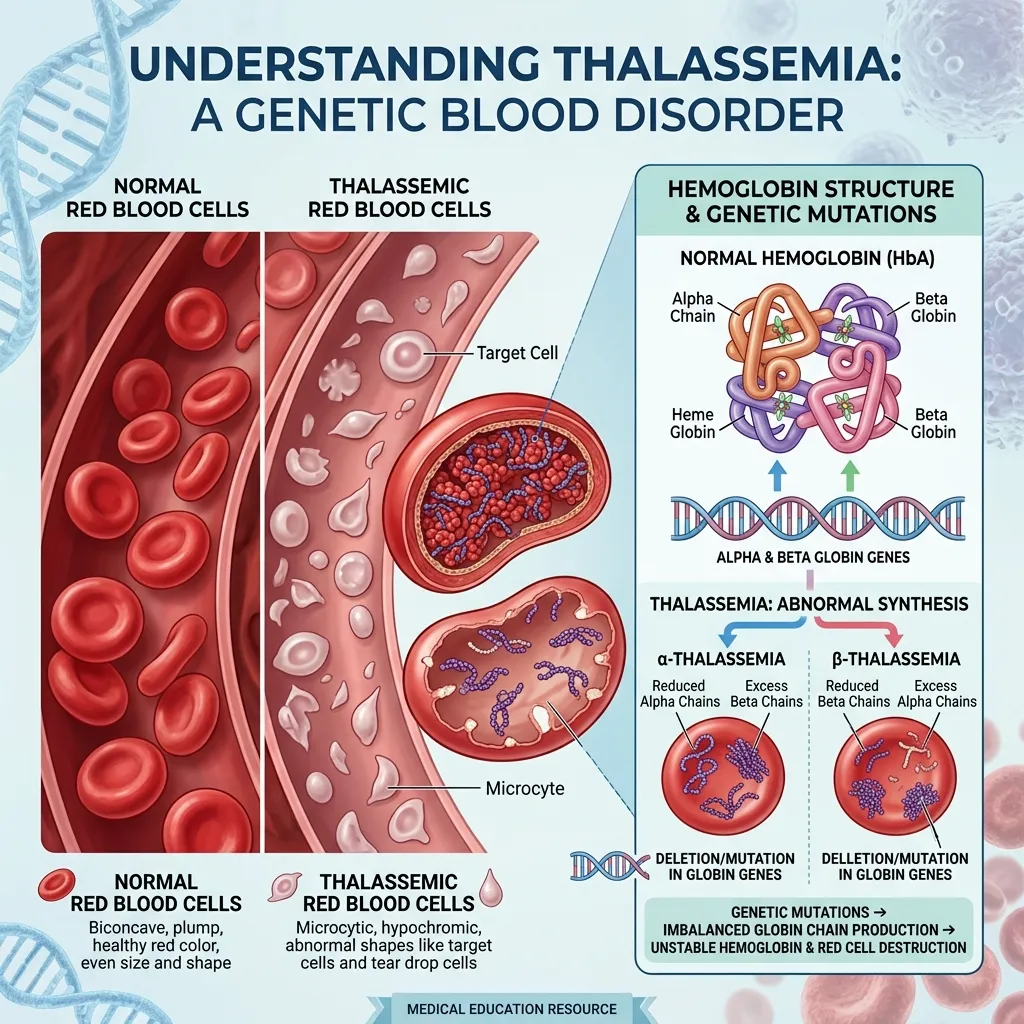 Thalassemia is a group of inherited blood disorders caused by reduced or absent production of the protein chains that make up hemoglobin. Hemoglobin is the molecule inside red blood cells that carries oxygen from your lungs to every tissue in your body. When hemoglobin production falters, the result is anemia that ranges from mild to life-threatening.
Thalassemia is a group of inherited blood disorders caused by reduced or absent production of the protein chains that make up hemoglobin. Hemoglobin is the molecule inside red blood cells that carries oxygen from your lungs to every tissue in your body. When hemoglobin production falters, the result is anemia that ranges from mild to life-threatening.
Healthy adult hemoglobin (HbA) is built from four protein chains: two alpha-globin chains and two beta-globin chains. This balanced structure keeps red blood cells stable and oxygen flowing efficiently. When a genetic change disrupts the production of either chain, the alpha-to-beta balance collapses, unstable hemoglobin forms, and red blood cells break down prematurely.
Why Does Understanding Inheritance Patterns Matter?
Understanding the thalassemia inheritance pattern is not just academic. It shapes diagnosis, prevention, treatment decisions, and family planning for millions of people worldwide. Because the condition is passed from parents to children, knowing your genetic status before starting a family carries real weight. According to the World Health Organization, expanded screening and genetic counseling can significantly reduce the number of children born with severe forms.
What Genetic Principles Are Relevant to Thalassemia?
Thalassemia follows the rules of classic genetics. The genes that build globin chains live on specific chromosomes, and you inherit one copy from each parent. Whether you stay healthy, become a silent carrier, or develop severe anemia depends on how many of these genes are affected and which combination you inherit. These principles form the backbone of thalassemia genetic inheritance, and the rest of this guide builds on them.
The Basics of Genetic Inheritance
Before diving into how thalassemia is inherited, it helps to understand a few core concepts that govern all genetic conditions.
Genes, Chromosomes, and DNA
Your DNA is the instruction manual for building and running your body. DNA is packaged into structures called chromosomes, and most people carry 23 pairs—46 chromosomes in total. Each chromosome holds thousands of genes, the specific segments of DNA that code for proteins. The genes responsible for thalassemia direct the production of alpha-globin and beta-globin chains. Alpha-globin genes sit on chromosome 16, while the beta-globin gene sits on chromosome 11.
Dominant vs. Recessive Traits
Genes come in different versions, and how those versions behave determines what traits you express. A dominant trait shows up even if you inherit just one copy of the gene variant. A recessive trait, by contrast, only appears when you inherit two affected copies—one from each parent. Thalassemia is a recessive condition, which is why a single affected gene often produces no symptoms at all.
Homozygous vs. Heterozygous States
When both copies of a gene are identical, you are homozygous for that gene. When the two copies differ, you are heterozygous. In thalassemia, someone who inherits one affected gene and one normal gene is heterozygous—usually a carrier with mild or no symptoms. Someone who inherits two affected genes is homozygous and typically develops a more severe form. This distinction is central to the thalassemia inheritance pattern.
Autosomal Recessive Inheritance: The Core Pattern of Thalassemia
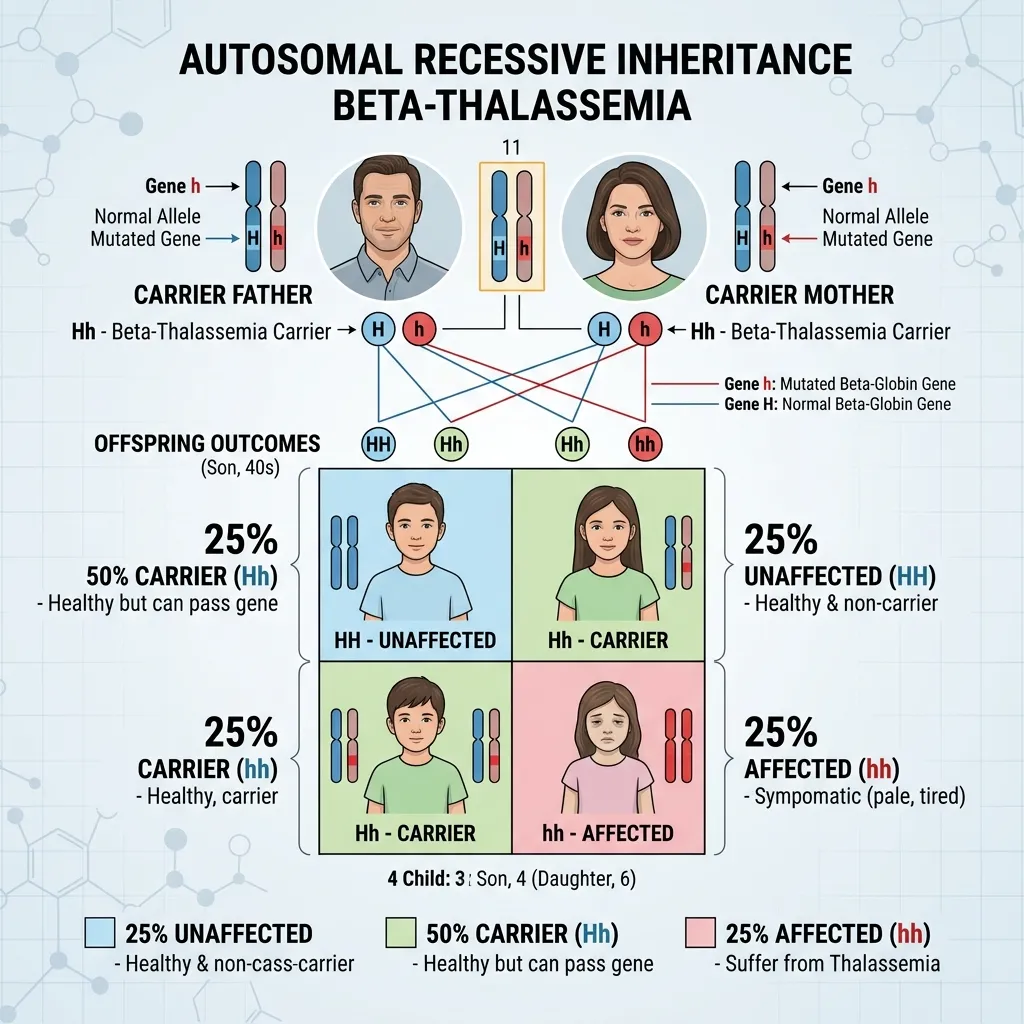 Every form of thalassemia shares one defining feature: it follows an autosomal recessive inheritance pattern. Grasping this single concept unlocks the entire picture.
Every form of thalassemia shares one defining feature: it follows an autosomal recessive inheritance pattern. Grasping this single concept unlocks the entire picture.
What Is Autosomal Recessive Inheritance?
Autosomal recessive inheritance means two things. “Autosomal” refers to the fact that the affected genes sit on the autosomes—the numbered chromosomes (1 through 22)—rather than the sex chromosomes (X and Y). This means thalassemia affects males and females equally. “Recessive” means a person must inherit two affected copies of the gene to develop the disease. One affected copy alone usually produces only carrier status.
How Is Thalassemia Inherited? A Detailed Explanation
When both parents are carriers of thalassemia, each pregnancy carries a predictable set of odds. Each parent passes on either their normal gene or their affected gene, entirely by chance.
This produces three possible outcomes for the child:
- 25% chance the child inherits two normal genes and is completely unaffected.
- 50% chance the child inherits one normal and one affected gene, becoming a carrier like the parents.
- 25% chance the child inherits two affected genes and develops a more severe form of the disease.
These odds apply to every single pregnancy independently. A family could have one affected child followed by three unaffected children, or any other combination. The percentages describe probability, not a guaranteed pattern within a single family. This is the heart of autosomal recessive inheritance thalassemia.
The Role of Carrier Status
Carriers—also called those with thalassemia trait or minor—are the silent engine of thalassemia genetic inheritance. They usually feel perfectly healthy, often with only mild anemia that can be mistaken for iron deficiency. Because they show few or no symptoms, many carriers remain undiagnosed until they have an affected child or undergo genetic screening. Two carriers who have children together are the most common route to a severe case, which is why carrier identification is so important.
Types of Thalassemia and Their Inheritance
While the autosomal recessive pattern applies across the board, the two main types—alpha and beta thalassemia—differ in the genes involved and the way severity unfolds.
Alpha Thalassemia Inheritance
Alpha thalassemia is driven mostly by the loss of alpha-globin genes, making its inheritance pattern a little more complex than beta thalassemia.
The Alpha-Globin Genes (HBA1 and HBA2)
Two genes produce alpha-globin chains: HBA1 and HBA2. Both sit on chromosome 16, close together, and produce identical proteins. Most people inherit four functional alpha-globin genes—two from each parent. Because both genes make the same protein, the total number of working genes matters more than which specific gene is affected. You can explore how these genes drive oxygen transport in our detailed guide on HBA1 and HBA2 gene function.
Deletion Mechanisms
Most cases of alpha thalassemia trace back to alpha-globin gene deletion—the physical loss of one or more genes from chromosome 16. These deletions usually occur through non-allelic homologous recombination during the cell division that creates eggs and sperm. Because HBA1 and HBA2 look so alike, chromosomes can misalign and swap genetic material unevenly, leaving one chromosome short a gene. Our breakdown of the alpha globin gene deletion mechanism explains exactly how this happens at the DNA level.
Carrier States: Silent Carrier and Alpha Thalassemia Trait
The number of affected alpha genes maps almost perfectly onto severity. When one of four genes is affected, the result is a silent carrier with normal hemoglobin and no symptoms, usually found only through genetic screening. When two genes are affected, the result is alpha thalassemia trait—mild microcytic anemia with small red blood cells, often mistaken for iron deficiency.
Hemoglobin H Disease
When three of the four alpha genes are affected, the result is Hemoglobin H disease. The severe shortage of alpha chains lets excess beta chains form unstable Hemoglobin H. Patients face moderate to severe hemolytic anemia, an enlarged spleen, and sometimes bone changes, requiring ongoing medical care.
Hydrops Fetalis
When all four alpha genes are affected, the result is Hemoglobin Bart’s hydrops fetalis—the most severe form. With no alpha-globin at all, the fetus produces Hemoglobin Bart’s, leading to massive fluid buildup and heart failure. This condition is usually fatal before or shortly after birth. It typically arises only when both parents carry a double-gene deletion on a single chromosome, a key reason molecular testing matters for at-risk couples.
Beta Thalassemia Inheritance
Beta thalassemia follows a more straightforward two-gene inheritance pattern, since a single gene controls beta-globin production.
The Beta-Globin Gene (HBB)
The beta-globin chain is produced by a single gene called HBB, located on chromosome 11. Each person inherits two copies—one from each parent. Unlike alpha thalassemia, which is mostly caused by deletions, beta thalassemia usually results from point mutations that reduce or abolish beta-globin production. The combination of mutations a person inherits determines where they land on the severity spectrum.
Carrier States: Beta Thalassemia Trait (Minor)
A person who inherits one normal HBB gene and one affected copy has beta thalassemia trait, also called beta thalassemia minor. These carriers usually have mild anemia or no symptoms at all. As with alpha thalassemia, the trait is frequently mistaken for iron deficiency, which can lead to unnecessary iron supplementation.
Thalassemia Intermedia
When both HBB genes are affected but some beta-globin production continues, the result is thalassemia intermedia. This intermediate form causes moderate anemia. Some patients need occasional blood transfusions, while others manage without regular transfusion support, depending on the specific mutations involved.
Thalassemia Major (Cooley’s Anemia)
When both HBB genes are severely affected and beta-globin production nearly stops, the result is beta thalassemia major, historically known as Cooley’s anemia. This is the most severe form of beta thalassemia. Children typically show signs within the first two years of life and require regular, lifelong blood transfusions along with iron chelation therapy to manage the iron overload that transfusions cause.
Genetic Testing and Counseling for Thalassemia

Because thalassemia is inherited, understanding your genetic status before starting a family gives you the widest range of choices.
When to Consider Testing
Testing is worth considering if you have a family history of anemia or thalassemia, if you belong to a population with high carrier rates—such as people of Southeast Asian, Mediterranean, Middle Eastern, or African descent—or if a routine blood test reveals unexplained microcytic anemia. Couples planning a family, especially when both partners may carry the trait, benefit most from early testing.
Types of Genetic Tests
Diagnosis usually follows a layered approach that moves from indirect clues to direct genetic evidence:
- Complete Blood Count (CBC): Often reveals microcytosis—red blood cells smaller than normal—and reduced MCH.
- Hemoglobin electrophoresis or HPLC: Detects abnormal hemoglobins and helps distinguish thalassemia types, though results can be normal in milder cases.
- Molecular genetic testing: Methods like Gap-PCR, MLPA (Multiplex Ligation-dependent Probe Amplification), and Next-Generation Sequencing directly analyze DNA to confirm specific deletions and mutations.
Genetic testing is the gold standard because it identifies the underlying cause directly. Choose molecular testing if confirming carrier status matters more to you than a quick preliminary screen—especially when both partners may carry affected genes.
The Importance of Genetic Counseling
Genetic counseling helps carriers understand their inheritance risk, interpret test results, and weigh their options without pressure. A counselor can map out the odds for each pregnancy, explain what the numbers mean in practical terms, and connect families with support resources. For couples who are both carriers, this guidance reduces the emotional and medical burden that a severe diagnosis can bring.
Family Planning and Risk Assessment
When both partners are carriers, several reproductive paths are available. These include prenatal diagnosis using chorionic villus sampling or amniocentesis, and IVF with preimplantation genetic testing, which screens embryos before implantation. Our family planning thalassemia guide covers carrier screening, prenatal diagnosis, and these reproductive choices in detail.
Living With Thalassemia: Implications of Inheritance
The thalassemia inheritance pattern shapes not just diagnosis but daily life—for patients, carriers, and entire families.
Impact on Individuals With Thalassemia
For people with severe forms like thalassemia major or Hemoglobin H disease, life often involves regular medical monitoring, transfusions, and ongoing care. Yet with modern treatment, many people with thalassemia live full, active lives. Those with milder forms, including the trait and silent carrier states, usually carry on with little to no disruption.
Impact on Carriers and Families
For carriers, the main implication is reproductive. A carrier’s health rarely suffers, but their genetic status becomes meaningful when planning a family. Learning you are a carrier can prompt important conversations with a partner and a healthcare provider. For broader context on how inherited blood conditions affect families, our genetic blood disorders guide connects the dots across related conditions.
Management and Treatment Options
Treatment depends entirely on severity. Silent carriers and those with the trait need no treatment, though they should avoid unnecessary iron supplements. Hemoglobin H disease and thalassemia intermedia may call for folic acid supplementation and occasional transfusions. Severe forms such as thalassemia major require regular blood transfusions and chelation therapy to remove excess iron and protect organs like the heart and liver. For the most severe cases, bone marrow or stem cell transplant offers a potential cure.
Future Directions in Thalassemia Research and Treatment
 The future of thalassemia care is moving fast, driven by advances in genetics and biotechnology.
The future of thalassemia care is moving fast, driven by advances in genetics and biotechnology.
Gene Therapy Advancements
Gene therapy offers the most promising path forward. Researchers are developing techniques to harvest a patient’s own hematopoietic stem cells, insert functional globin genes, and transplant them back—potentially curing the condition without lifelong transfusions. Gene-editing tools like CRISPR may expand these possibilities even further, aiming to correct the underlying genetic defect directly.
New Diagnostic Techniques
Next-Generation Sequencing is making it easier and faster to identify rare mutations and tailor treatment to each patient’s exact genetic profile. As these molecular tools become more affordable and accessible, carrier screening and prenatal diagnosis will reach more families, helping to reduce the number of children born with severe forms.
Progress in gene therapy and molecular diagnostics offers genuine hope. With knowledge, screening, and the right support, families affected by thalassemia can face the future with confidence.








{kind=link}