

")
Every breath you take depends on a tiny set of genetic instructions working perfectly. Two genes—HBA1 and HBA2—sit quietly on chromosome 16, telling your body how to build alpha-globin chains. These chains are the backbone of hemoglobin, the protein that moves oxygen from your lungs to every cell in your body.
When HBA1 and HBA2 gene function works as intended, your blood carries oxygen efficiently and your red blood cells stay healthy. When something goes wrong—a deletion, a point mutation, a structural error—the balance breaks down. The result can range from no symptoms at all to severe, life-threatening anemia.
This guide explains how the HBA1 and HBA2 genes in hemoglobin work, where they sit on the genome, and how they keep oxygen flowing. You’ll also learn how alpha thalassemia gene mutations disrupt this process, how doctors diagnose the resulting conditions, and what treatments and research are shaping the future. Whether you’re a student, a healthcare professional, or someone with a family history of anemia, this is your foundation for understanding alpha globin gene function.
What Is Hemoglobin and Why Does It Matter?
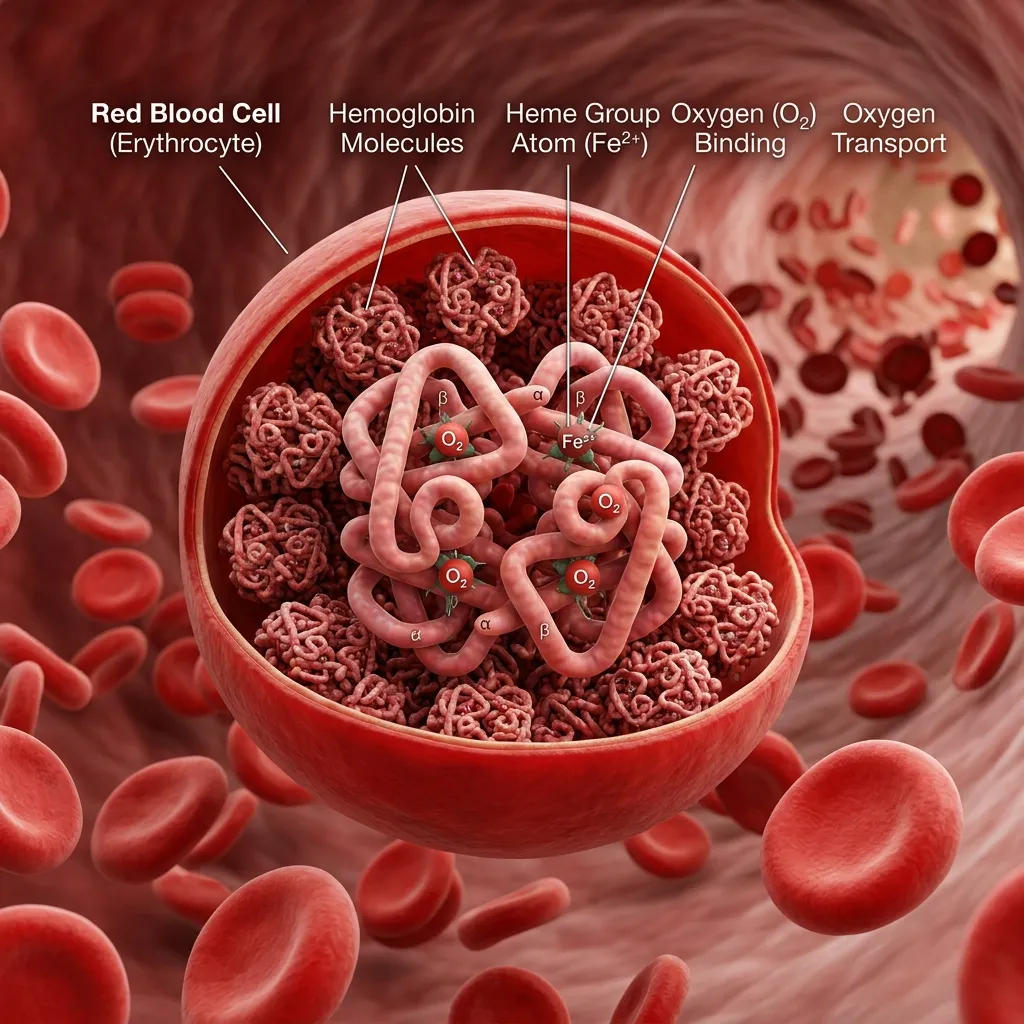 Hemoglobin is the protein inside red blood cells that binds oxygen in the lungs and delivers it throughout the body. Without it, your tissues and organs would starve for oxygen within minutes.
Hemoglobin is the protein inside red blood cells that binds oxygen in the lungs and delivers it throughout the body. Without it, your tissues and organs would starve for oxygen within minutes.
Healthy adult hemoglobin, called Hemoglobin A (HbA), is built from four protein chains: two alpha-globin chains and two beta-globin chains. This balanced four-part structure keeps red blood cells stable and oxygen delivery smooth. HbA makes up over 95% of the hemoglobin in adults.
The Role of Alpha-Globin Chains
Alpha-globin chains are essential building blocks of hemoglobin. They must pair with beta-globin chains in equal amounts to form stable HbA molecules. This balance—an even ratio of alpha to beta chains—is the secret to healthy red blood cells.
When alpha-globin production drops, beta chains in adults (or gamma chains in fetuses) are left unpaired. These leftover chains clump together into unstable structures that damage red blood cell membranes, leading to premature cell destruction and chronic anemia. This imbalance sits at the very heart of alpha thalassemia.
Introducing HBA1 and HBA2
HBA1 and HBA2 are the two genes responsible for producing alpha-globin chains. Both genes live on chromosome 16, sit very close to one another, and produce identical alpha-globin proteins. Most people inherit four functional alpha-globin genes—two from each parent—giving the body the steady supply of alpha chains it needs. For a closer look at how the underlying genetics drive disease, our guide on the molecular basis of alpha thalassemia breaks it down at the cellular level.
How Do the HBA1 and HBA2 Genes Work?
The HBA1 and HBA2 gene function activates during red blood cell formation in the bone marrow. These genes instruct cells to produce alpha-globin chains, which then combine with beta-globin chains to form functional hemoglobin.
The process follows a clear sequence:
- The genes activate in the bone marrow during red blood cell production.
- They direct the synthesis of alpha-globin chains.
- These alpha chains combine with beta-globin chains.
- Together they form stable Hemoglobin A molecules.
- The finished hemoglobin transports oxygen through the bloodstream.
Where Are HBA1 and HBA2 Located on Chromosome 16?
The alpha-globin gene cluster sits on the short arm of chromosome 16, near the telomere at position 16p13.3. This region is highly active during genetic recombination, which is exactly why it becomes a hotspot for deletions.
HBA1 and HBA2 share nearly identical DNA sequences. That similarity is helpful for steady alpha-globin production, but it carries a hidden risk. Because the two genes look so alike, chromosomes can misalign during cell division—setting the stage for the deletions behind alpha thalassemia.
What Is the HBA1 Gene’s Role in Hemoglobin Production?
The HBA1 gene’s role in hemoglobin production is to ensure the continuous synthesis of alpha-globin chains throughout life, from early development into adulthood. It works in coordination with the other globin genes to keep hemoglobin production balanced, so red blood cells form properly and deliver oxygen efficiently.
When the HBA1 gene functions correctly, hemoglobin remains structurally stable, circulation runs smoothly, and energy levels stay normal. When mutations or deletions strike the HBA1 gene, alpha-globin production drops, hemoglobin becomes unstable, and red blood cells break down more easily—often leading to mild or severe alpha thalassemia depending on how many gene copies are affected.
What Are the Effects of HBA2 Gene Mutations?
The HBA2 gene also produces alpha-globin chains, so any mutation or deletion here reduces the overall alpha-globin supply. When HBA2 function is impaired, hemoglobin synthesis declines, unstable hemoglobin molecules form, and red blood cells become more fragile.
The effects grow more serious when both HBA1 and HBA2 are affected at the same time. The drop in alpha-globin production becomes far more pronounced, sharply increasing the risk of significant alpha thalassemia that may require long-term medical care.
How Does Alpha Globin Gene Function Drive Oxygen Transport?
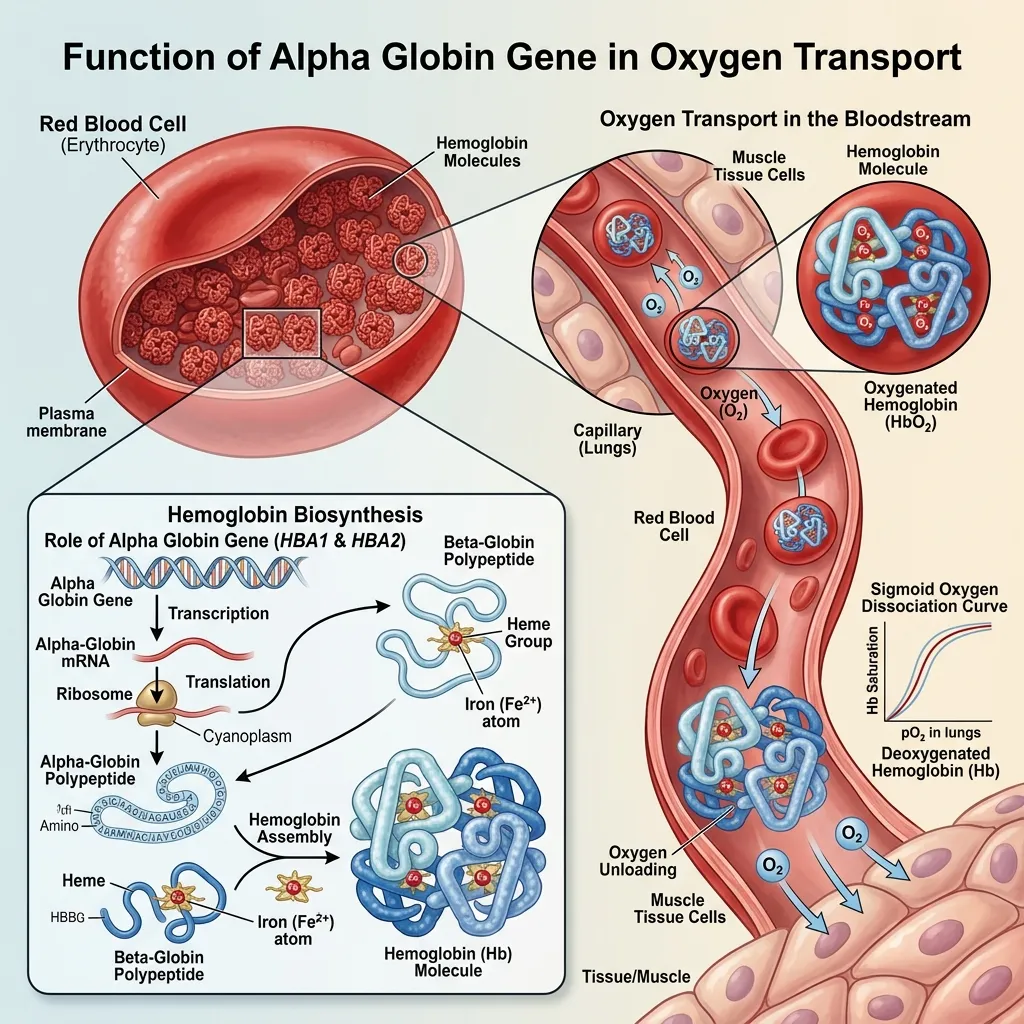 The alpha globin gene function in hemoglobin synthesis is to produce alpha chains in equal amounts to beta chains. This one-to-one balance is what makes hemoglobin stable enough to carry oxygen reliably.
The alpha globin gene function in hemoglobin synthesis is to produce alpha chains in equal amounts to beta chains. This one-to-one balance is what makes hemoglobin stable enough to carry oxygen reliably.
When alpha-globin production falls short, a chain reaction begins:
- Excess beta chains accumulate without partners.
- Abnormal, unstable hemoglobin forms.
- Red blood cells lose their structural integrity.
- Oxygen delivery to tissues declines.
This breakdown is the central cause of alpha thalassemia and several related genetic blood disorders. Understanding the role of HBA1 and HBA2 genes in hemoglobin helps explain why even small genetic changes can have outsized effects on oxygen transport and overall health.
What Genetic Variations and Mutations Affect HBA1 and HBA2?
Alpha thalassemia gene mutations come in two broad categories: deletions and non-deletion mutations. Both reduce alpha-globin production, but they do so in different ways.
Deletional Mutations
Deletions are the most common cause of alpha thalassemia. They involve the physical loss of one or more alpha-globin genes from chromosome 16. Most occur through non-allelic homologous recombination during meiosis—the cell division that creates eggs and sperm. Because HBA1 and HBA2 look so similar, chromosomes can misalign and swap genetic material unevenly, leaving one chromosome short a gene. Our detailed breakdown of the alpha globin gene deletion mechanism explains exactly how this happens.
Non-Deletional Mutations
A smaller share of cases come from non-deletion mutations, where the gene stays physically present but stops working properly. These include point mutations (single base-pair changes), splice site mutations, and mutations in regulatory regions. Unlike full deletions, these often allow partial alpha-globin production, which means disease severity can vary widely.
How Many Genes Are Affected Determines Severity
The number of working alpha-globin genes maps directly onto disease severity—one of the most predictable patterns in genetic medicine:
- One gene affected: Silent carrier with no symptoms.
- Two genes affected: Alpha thalassemia trait, with mild anemia and small red blood cells.
- Three genes affected: Hemoglobin H disease, with moderate to severe anemia.
- Four genes affected: Hemoglobin Bart’s hydrops fetalis, usually fatal before or shortly after birth.
What Is Alpha Thalassemia and How Is It Diagnosed?
Alpha thalassemia is a group of inherited blood disorders caused by reduced or absent alpha-globin production. Because HBA1 and HBA2 produce identical proteins, the total count of working genes matters more than which specific gene is lost.
The Spectrum of Alpha Thalassemia Syndromes
Doctors classify alpha thalassemia into four syndromes based on how many alpha-globin genes are missing or defective:
- Silent carrier (one gene): Normal hemoglobin, no symptoms, usually found only through genetic screening.
- Alpha thalassemia trait (two genes): Mild microcytic anemia, often mistaken for iron deficiency. Our complete guide to the alpha thalassemia trait covers testing and care in detail.
- Hemoglobin H disease (three genes): Moderate to severe hemolytic anemia, an enlarged spleen, and sometimes bone changes.
- Hydrops fetalis (four genes): The most severe form, producing Hemoglobin Bart’s and usually fatal in the womb or shortly after birth.
Diagnostic Methods for Alpha Thalassemia
Standard blood tests can hint at alpha thalassemia, but they rarely confirm it. Diagnosis usually follows a layered approach:
- Complete Blood Count (CBC): Often reveals microcytosis—red blood cells smaller than normal—and reduced MCH.
- Hemoglobin electrophoresis or HPLC: May detect abnormal hemoglobins like Hb H or Hemoglobin Bart’s, though results are often normal in milder cases.
- Molecular genetic testing: Methods like Gap-PCR, MLPA, and Next-Generation Sequencing directly analyze DNA to confirm specific deletions and mutations.
Genetic testing is considered the gold standard because it identifies the underlying cause directly rather than relying on indirect blood patterns.
Why Early Detection Matters
Accurate, early diagnosis prevents unnecessary iron supplementation in carriers whose anemia is genetic, not iron-related. It also gives at-risk families the time and knowledge to make informed decisions about screening and family planning—especially when both partners may carry deletions.
How Is Alpha Thalassemia Treated and Managed?
 Treatment depends entirely on severity, which the deletion count predicts. There is no need for a one-size-fits-all approach—care is tailored to the individual.
Treatment depends entirely on severity, which the deletion count predicts. There is no need for a one-size-fits-all approach—care is tailored to the individual.
Current Treatment Approaches
- Silent carriers and trait: No treatment needed. These individuals should avoid unnecessary iron supplements, since their anemia is genetic rather than iron-related.
- Hemoglobin H disease: Care often includes folic acid supplementation, periodic blood transfusions during anemic crises, and sometimes splenectomy to extend the lifespan of red blood cells.
- Hydrops fetalis: Historically fatal, this condition can occasionally be managed with intrauterine transfusions, followed by lifelong transfusion therapy if the infant survives.
Emerging Therapies and Research
Gene therapy offers the most promising path forward. Researchers are developing techniques to harvest a patient’s own hematopoietic stem cells, insert functional alpha-globin genes, and transplant them back—potentially curing the condition without lifelong transfusions. Gene-editing tools like CRISPR may expand these possibilities even further. Next-Generation Sequencing is also making it easier to identify rare mutations and tailor treatment.
Genetic Counseling and Family Planning
Genetic counseling is vital for at-risk families. Couples who are both carriers face a higher chance of having a child with a severe form of alpha thalassemia. Our family planning thalassemia guide covers carrier screening, prenatal diagnosis, and reproductive options like IVF with preimplantation genetic testing.
Why Does HBA1 and HBA2 Research Matter?
Understanding HBA1 and HBA2 gene function reaches far beyond a single diagnosis. It shapes hematology, public health, and the future of genetic medicine.
In clinical genetics, knowing the precise effects of HBA1 and HBA2 mutations supports early diagnosis of genetic anemia, smarter family planning decisions, and accurate prenatal screening. For a wider view of how gene mutations affect blood health, our genetic blood disorders guide connects the dots across conditions.
On a population level, alpha thalassemia is one of the most common inherited blood disorders worldwide, with high carrier rates in Southeast Asia, the Mediterranean, the Middle East, and parts of Africa. According to the World Health Organization, expanded screening and genetic counseling can significantly reduce the number of children born with severe forms. The Centers for Disease Control and Prevention offers similar trusted guidance on screening and care.
Looking ahead, advances in gene therapy, faster molecular diagnostics, and personalized medicine are transforming how these disorders are managed. The more precisely we understand each patient’s HBA1 and HBA2 gene function, the better hematologists can predict severity, tailor treatment, and protect organs from long-term damage.
Conclusion
The HBA1 and HBA2 gene function may seem simple, but these genes perform one of the most important tasks in human biology: producing the alpha-globin chains required for healthy hemoglobin. Understanding alpha globin gene function helps explain how red blood cells transport oxygen efficiently throughout the body. The close relationship between HBA1 and HBA2 genes in hemoglobin production ensures that tissues receive the oxygen needed for normal growth, metabolism, and survival.
When mutations or deletions affect these genes, alpha-globin production decreases, leading to alpha thalassemia gene mutations and a range of blood disorders. Depending on how many genes are affected, the condition can vary from a silent carrier state to severe, life-threatening anemia. Early detection and genetic testing are therefore essential for accurate diagnosis and effective disease management.
Frequently Asked Questions
1. What is HBA1 and HBA2 gene function?
HBA1 and HBA2 genes produce alpha-globin chains, essential components of hemoglobin. These chains combine with beta-globin chains to form functional hemoglobin in red blood cells, ensuring efficient oxygen transport throughout the body.
2. Where are the HBA1 and HBA2 genes located?
Both genes sit on the short arm of chromosome 16 at position 16p13.3, very close to each other. This proximity supports balanced alpha-globin production but also makes the region prone to deletions.
3. What is the HBA1 gene’s role in hemoglobin production?
The HBA1 gene ensures continuous synthesis of alpha-globin chains throughout life. It supports stable hemoglobin structure and efficient oxygen delivery, working in coordination with HBA2 and the beta-globin genes.
4. What happens when the HBA2 gene mutates?
A mutation or deletion in the HBA2 gene reduces alpha-globin production, leading to unstable hemoglobin and lower oxygen-carrying capacity. Effects are more severe when both HBA1 and HBA2 are affected together.
5. How do alpha thalassemia gene mutations cause disease?
Alpha thalassemia gene mutations—whether deletions or point mutations—reduce alpha-globin production. This creates an imbalance between alpha and beta chains, producing unstable hemoglobin, fragile red blood cells, and chronic anemia.
6. How many alpha-globin genes do people normally have?
Most people inherit four functional alpha-globin genes—two HBA1 and two HBA2 copies—one pair from each parent. The number of working genes determines the severity of any resulting alpha thalassemia.
7. How is alpha thalassemia diagnosed?
Diagnosis usually starts with a Complete Blood Count showing small red blood cells, followed by hemoglobin analysis. Molecular genetic testing, such as Gap-PCR or MLPA, provides definitive confirmation of specific deletions or mutations.
8. Can alpha thalassemia be cured?
Most forms cannot be cured today, though symptoms can be managed with monitoring, transfusions, and folic acid. Severe cases may be treated with stem cell transplant, and experimental gene therapy shows strong future promise.
9. Is alpha thalassemia inherited?
Yes. Alpha thalassemia follows an autosomal recessive pattern. A child must inherit affected genes from both parents to develop a severe form, which is why carrier screening matters for couples planning a family.
10. Why is understanding HBA1 and HBA2 gene function important?
These genes are critical for oxygen transport and overall blood health. Understanding their function supports early diagnosis, accurate genetic counseling, prenatal screening, and broader prevention of inherited blood disorders.








{kind=link}