


The alpha globin gene deletion mechanism primarily occurs through non-allelic homologous recombination (NAHR) during meiosis. This unequal crossing over results in the loss of HBA1 or HBA2 genes on chromosome 16. The severity of the resulting alpha-thalassemia directly correlates with the number of deleted alpha-globin genes.
Alpha-thalassemia is a highly prevalent inherited blood disorder characterized by the reduced or absent production of alpha-globin chains. These chains are essential building blocks of hemoglobin, the protein responsible for carrying oxygen throughout the human body. Understanding the precise molecular events that trigger this condition is crucial for medical professionals, genetic counselors, and affected families.
The core of this condition lies in the alpha globin gene deletion mechanism. Unlike some genetic disorders caused primarily by single nucleotide changes, alpha-thalassemia is predominantly driven by the physical loss of genetic material. This loss disrupts the delicate balance of hemoglobin chain synthesis, leading to the destruction of red blood cells and varying degrees of anemia.
By exploring the specific alpha thalassemia gene mutation process, researchers have uncovered how genetic misalignments during cellular division lead to these deletions. This blog post will detail the fundamental mechanisms behind these gene deletions, the specific roles of the HBA1 and HBA2 genes, and the profound clinical implications of a chromosome 16 globin gene defect. You will learn how these genetic alterations manifest in patients and the steps medical science is taking to diagnose and manage them.
How do the HBA1 and HBA2 genes function in the body?
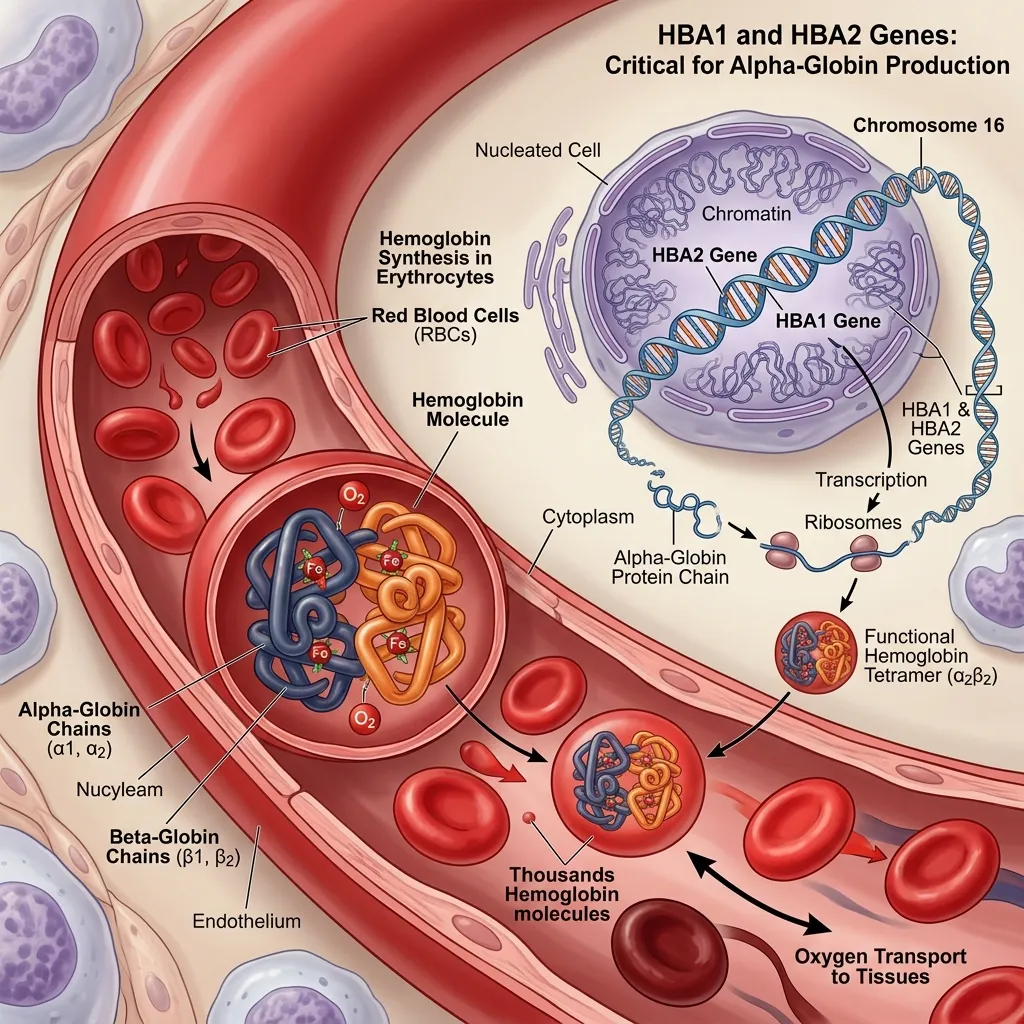 The human body relies on a precise genetic blueprint to produce functional hemoglobin. The instructions for manufacturing the alpha-globin subunit of hemoglobin are housed within the alpha-globin gene cluster.
The human body relies on a precise genetic blueprint to produce functional hemoglobin. The instructions for manufacturing the alpha-globin subunit of hemoglobin are housed within the alpha-globin gene cluster.
What is the structure of the alpha-globin genes?
Humans normally inherit four functional alpha-globin genes. You receive two genes from your mother and two from your father. These specific genes are identified as HBA1 and HBA2. They are positioned closely together on the short arm of chromosome 16. According to the Centers for Disease Control and Prevention, 2024] (https://www.cdc.gov/ncbddd/thalassemia/index.html), the proximity and high sequence similarity of HBA1 and HBA2 play a significant role in their susceptibility to genetic mutations.
How do HBA1 and HBA2 contribute to normal hemoglobin production?
The HBA1 and HBA2 genes share identical coding sequences and produce identical alpha-globin protein chains. These alpha-globin chains must pair with beta-globin chains to form Hemoglobin A (HbA), which comprises over 95% of adult hemoglobin. A healthy individual requires a steady, equal production of both alpha and beta chains to maintain stable red blood cells.
What are the consequences of altered HBA1 and HBA2 activity?
When the alpha globin gene deletion mechanism removes one or more of these genes, the body produces fewer alpha-globin chains. This creates an excess of beta-globin chains in adults, or gamma-globin chains in fetuses. These unpaired chains form unstable tetramers that damage the red blood cell membrane, leading to premature cell death and anemia. The specific HBA1 HBA2 gene deletion effects dictate the clinical severity of the patient’s condition.
What drives the alpha-globin gene deletion mechanisms?
The alpha thalassemia gene mutation process is heavily influenced by the structural layout of chromosome 16. Because the HBA1 and HBA2 genes are highly homologous (similar in DNA sequence) and located near each other, they are prone to structural rearrangements.
How does unequal crossing over lead to gene deletion?
The most frequent alpha globin gene deletion mechanism is a process called unequal crossing over. During meiosis—the cellular division that creates sperm and egg cells—homologous chromosomes align to exchange genetic information. Because the alpha-globin gene cluster contains repetitive DNA sequences known as Z boxes and X boxes, the chromosomes can easily misalign.
What is Non-allelic Homologous Recombination (NAHR)?
When these misaligned chromosomes exchange genetic material, it triggers Non-allelic Homologous Recombination (NAHR). According to genetic researchers, NAHR results in one chromosome gaining extra genetic material (a duplication) while the other chromosome loses genetic material (a deletion). The chromosome bearing the deletion will carry the chromosome 16 globin gene defect into the next generation.
What are the most common deletion types?
Medical professionals categorize these deletions based on the specific segments of DNA lost. Common deletions include the -alpha3.7 and -alpha4.2 single-gene deletions, which leave one functional alpha gene on the affected chromosome. Larger structural losses, such as the –SEA (Southeast Asian) or –MED (Mediterranean) deletions, remove both HBA1 and HBA2 genes from a single chromosome, leading to more severe clinical outcomes.
What other deletion mechanisms exist?
While unequal crossing over accounts for the vast majority of alpha-thalassemia cases, the alpha thalassemia gene mutation process can also involve less common genetic events.
How do point mutations cause alpha-thalassemia?
In some populations, non-deletional alpha-thalassemia occurs due to point mutations. These are single base-pair changes within the HBA1 or HBA2 genes that prevent the gene from producing a functional protein, even though the gene itself remains physically present on the chromosome.
Are insertions and inversions common?
Insertions and inversions are rare structural variations that disrupt the regulatory elements controlling the alpha-globin genes. While infrequent, they still result in impaired alpha-globin production and contribute to the spectrum of alpha-thalassemia disorders.
What are the clinical effects of the alpha-thalassemia mutation process?
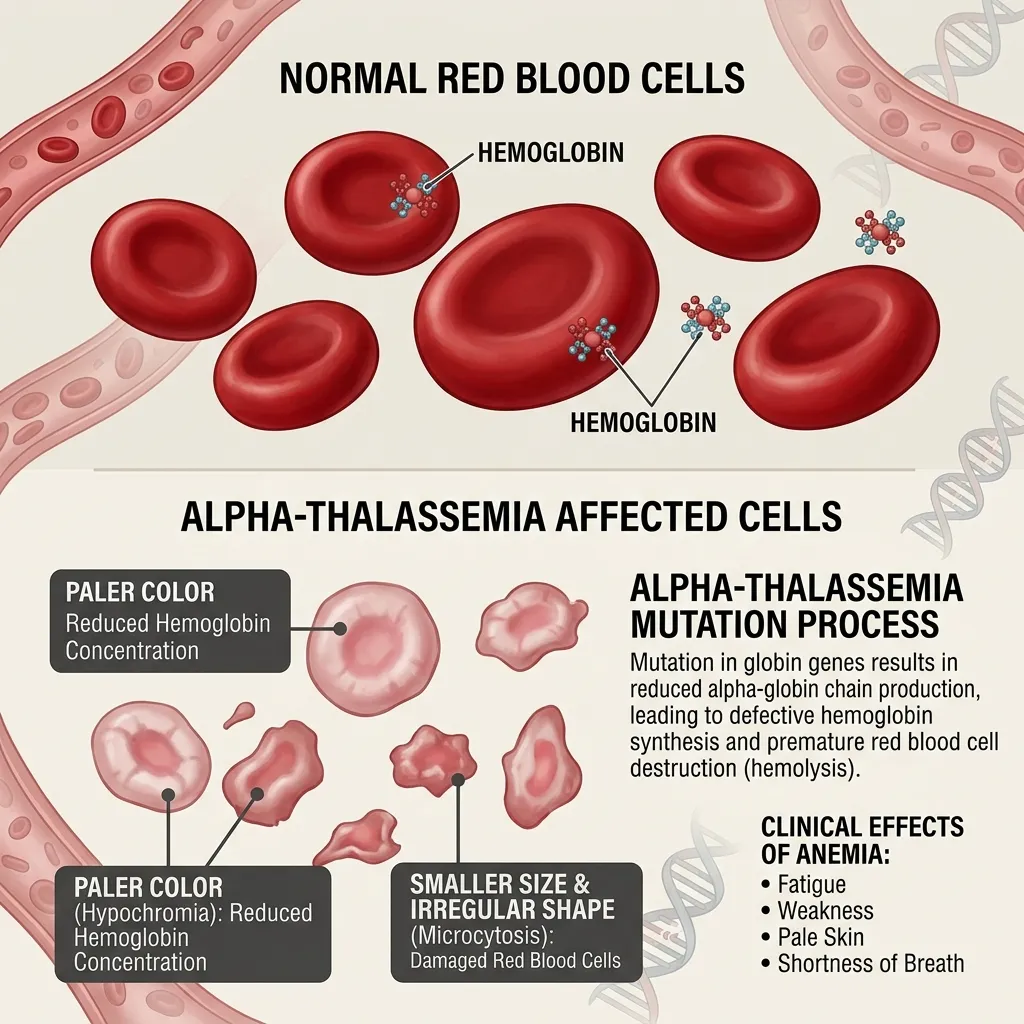 The HBA1 HBA2 gene deletion effects manifest across a wide clinical spectrum, directly corresponding to the number of deleted genes.
The HBA1 HBA2 gene deletion effects manifest across a wide clinical spectrum, directly corresponding to the number of deleted genes.
How does the genotype correlate with the physical phenotype?
Medical practitioners classify alpha-thalassemia into four distinct clinical syndromes based on how many of the four total alpha-globin genes are missing.
What happens if you are a silent carrier?
A silent carrier has lost one functional alpha-globin gene (-a/aa). These individuals typically exhibit normal hemoglobin levels and experience no physical symptoms. The condition is often only discovered during targeted genetic screening.
What are the symptoms of the alpha-thalassemia trait?
When a patient loses two alpha-globin genes, they exhibit the alpha-thalassemia trait. This can occur either through the deletion of one gene on each chromosome (-a/-a) or two genes on the same chromosome (–/aa). Patients typically present with mild microcytic anemia. For more details on managing this carrier state, you can read this comprehensive guide on the alpha thalassemia trait.
What is Hb H Disease?
Hb H disease arises when three alpha-globin genes are deleted (–/-a). The severe lack of alpha-globin causes excess beta-globin chains to form Hemoglobin H. Patients suffer from moderate to severe hemolytic anemia, enlarged spleens, and bone deformities, requiring dedicated medical intervention.
What causes Hydrops Fetalis?
The most devastating chromosome 16 globin gene defect occurs when all four alpha-globin genes are deleted (–/–). Fetuses cannot produce any alpha-globin, resulting in Hemoglobin Barts. This condition causes massive fluid accumulation, heart failure, and is typically fatal before or shortly after birth. Expectant parents can learn more about this severe condition by reviewing this resource on understanding Hemoglobin Barts.
How do doctors diagnose these clinical manifestations?
Diagnosis relies heavily on routine Complete Blood Counts (CBC), which reveal reduced red blood cell size (microcytosis). Hemoglobin electrophoresis or high-performance liquid chromatography (HPLC) can detect abnormal hemoglobins like Hb H or Hemoglobin Barts. However, definitive diagnosis requires molecular genetic testing to identify the exact alpha globin gene deletion mechanism.
How does the chromosome 16 globin gene defect impact populations?
The distribution of the chromosome 16 globin gene defect is not random. It is closely tied to evolutionary history and geographic geography.
Where is the alpha-globin cluster located?
The alpha-globin gene cluster is located near the telomere of the short arm of chromosome 16 (16p13.3). This specific location is highly active during genetic recombination, making it a hotspot for the alpha globin gene deletion mechanism.
What are the genetic inheritance patterns?
Alpha-thalassemia follows an autosomal recessive inheritance pattern. A child must inherit a chromosome 16 globin gene defect from both parents to develop severe forms like Hb H disease or Hydrops Fetalis.
How does geographic distribution affect population genetics?
According to the [World Health Organization, 2023] (https://www.who.int/health-topics/thalassaemia), alpha-thalassemia is highly concentrated in populations from Southeast Asia, the Mediterranean, Africa, and the Middle East. Evolutionary biologists believe that carrying a single or double gene deletion historically provided some protection against severe malaria, causing these mutations to thrive in endemic regions.
What are the best diagnostic and screening approaches?
 Accurate identification of the alpha thalassemia gene mutation process is vital for patient care and family planning.
Accurate identification of the alpha thalassemia gene mutation process is vital for patient care and family planning.
Why is molecular genetic testing essential?
Standard blood tests indicate the presence of anemia but cannot confirm the specific HBA1 HBA2 gene deletion effects. Molecular genetic testing, specifically Polymerase Chain Reaction (PCR) and multiplex ligation-dependent probe amplification (MLPA), directly maps the patient’s DNA to locate exact deletions and point mutations. Choose molecular testing if definitive confirmation of carrier status matters more to you than a preliminary CBC screening.
How does prenatal diagnosis work?
For couples at risk of passing on severe chromosome 16 globin gene defects, prenatal diagnosis is critical. Doctors use chorionic villus sampling (CVS) or amniocentesis to extract fetal DNA. Analyzing this DNA allows medical teams to determine if the fetus suffers from Hydrops Fetalis. If you are exploring reproductive options, read this detailed family planning thalassemia guide.
What role does newborn screening play?
Newborn screening programs often test for the presence of Hemoglobin Barts in a baby’s blood. High levels of Hemoglobin Barts in a newborn strongly indicate a multi-gene deletion, allowing pediatricians to initiate monitoring and care immediately.
What management and treatment options exist for patients?
Treatment protocols are highly individualized, depending on the severity of the HBA1 HBA2 gene deletion effects.
How do doctors manage silent carriers and thalassemia traits?
Individuals missing one or two alpha-globin genes require no specific medical treatment. They should avoid unnecessary iron supplements, as their microcytic anemia is caused by genetics, not iron deficiency.
What are the treatments for Hb H Disease?
Patients missing three genes require ongoing hematological care. Treatment often includes folic acid supplementation to support red blood cell production, periodic blood transfusions during severe anemic crises, and occasionally splenectomy (removal of the spleen) to prolong the lifespan of red blood cells. To explore how this fits into the broader spectrum of genetic anemias, consult this genetic blood disorders guide.
How is Hydrops Fetalis treated?
Historically uniformly fatal, Hydrops Fetalis can now occasionally be managed with intrauterine blood transfusions. If the infant survives to term, they will require lifelong blood transfusions and intensive medical care, similar to patients with beta-thalassemia major.
What future directions and research are underway?
Medical science continues to push the boundaries of how we treat the alpha thalassemia gene mutation process.
Will gene therapy cure alpha-thalassemia?
Gene therapy represents the most promising future treatment. Researchers are developing techniques to harvest a patient’s own hematopoietic stem cells, insert functional copies of the alpha-globin genes, and transplant them back into the body. This could effectively cure the chromosome 16 globin gene defect without the need for lifelong transfusions.
What new diagnostic technologies are emerging?
Next-generation sequencing (NGS) is becoming more accessible, allowing laboratories to sequence the entire alpha-globin cluster rapidly. This technology accurately identifies rare non-deletional mutations and complex structural rearrangements that older tests might miss.
How does personalized medicine apply to thalassemia?
Personalized medicine approaches analyze a patient’s exact genetic profile to predict disease severity and tailor transfusion regimens. By understanding the precise HBA1 HBA2 gene deletion effects for an individual, hematologists can optimize chelation therapy and minimize organ damage.
Why is understanding the alpha globin gene deletion mechanism critical?
 The alpha globin gene deletion mechanism is a complex yet highly predictable biological process driven by the unique structure of chromosome 16. Unequal crossing over and non-allelic homologous recombination strip the body of the essential HBA1 and HBA2 genes, leading to a spectrum of physical ailments ranging from asymptomatic carrier states to fatal fetal hydrops.
The alpha globin gene deletion mechanism is a complex yet highly predictable biological process driven by the unique structure of chromosome 16. Unequal crossing over and non-allelic homologous recombination strip the body of the essential HBA1 and HBA2 genes, leading to a spectrum of physical ailments ranging from asymptomatic carrier states to fatal fetal hydrops.
Grasping the molecular realities of the alpha thalassemia gene mutation process allows healthcare providers to offer accurate diagnoses, precise genetic counseling, and life-saving interventions. As gene therapy and advanced molecular diagnostics continue to evolve, the medical community moves closer to eradicating the severe consequences of the chromosome 16 globin gene defect. If you or your partner has a family history of anemia, consult a hematologist or genetic counselor to undergo comprehensive genetic screening.
1. What is the main cause of alpha-thalassemia?
The main cause of alpha-thalassemia is the deletion or mutation of one or more of the four alpha-globin genes located on chromosome 16, which impairs the body’s ability to produce adequate hemoglobin.
2. How do HBA1 and HBA2 genes relate to alpha-thalassemia?
HBA1 and HBA2 are the specific genes responsible for instructing the body to make alpha-globin protein chains. The physical loss or mutation of these genes directly causes alpha-thalassemia.
3. Can alpha-thalassemia be cured?
Currently, severe alpha-thalassemia can only be cured through a successful bone marrow or stem cell transplant. However, experimental gene therapies are showing strong potential for future genetic cures.
4. What are the symptoms of the alpha-thalassemia trait?
Individuals with the alpha-thalassemia trait (missing two genes) typically experience mild microcytic anemia, which may cause slight fatigue, but most remain entirely asymptomatic.
5. How is alpha-thalassemia diagnosed?
Alpha-thalassemia is preliminarily detected through a Complete Blood Count (CBC) showing small red blood cells, but definitive diagnosis requires molecular DNA testing to identify the exact missing genes.
6. Is alpha-thalassemia inherited?
Yes. Alpha-thalassemia is a genetic condition inherited in an autosomal recessive pattern, meaning the defective genes are passed down from parents to their children.
7. What is unequal crossing over in the context of gene deletions?
Unequal crossing over is a cellular error during meiosis where misaligned chromosomes exchange genetic material unevenly, leading to one chromosome losing a gene (deletion) and the other gaining one (duplication).
8. What is the impact of a chromosome 16 globin gene defect?
A defect on chromosome 16 impairs the production of alpha-globin chains, leading to an imbalance in hemoglobin structure, premature destruction of red blood cells, and varying degrees of chronic anemia.
9. Are there different types of alpha-globin gene deletions?
Yes. Deletions range from single-gene losses (like -alpha3.7) to large, multi-gene deletions (like –SEA) that remove entire sections of the alpha-globin gene cluster.
10. Where can I find more information about alpha-thalassemia?
You can find comprehensive information through global health organizations like the CDC and WHO, or by consulting a certified genetic counselor and specialized hematology clinics.








{kind=link}