


Without proper HBA1 and HBA2 gene function, the body cannot produce normal hemoglobin, leading to disorders such as alpha thalassemia and other inherited blood diseases. These genes play a critical role in maintaining oxygen balance, energy production, and overall cellular health.
Understanding the HBA1 and HBA2 gene function is essential for students, researchers, and individuals studying genetic blood disorders because even small mutations in these genes can significantly impact health.
What Are HBA1 and HBA2 Genes?
The HBA1 and HBA2 gene function is directly related to the production of alpha globin chains. Both genes are located on chromosome 16 and work together to produce identical alpha globin proteins.
- HBA1 gene produces one form of alpha globin
- HBA2 gene produces another identical form of alpha globin
Although they produce similar proteins, both genes are necessary for maintaining balanced hemoglobin production.
The HBA1 gene’s role in hemoglobin production is to ensure continuous synthesis of alpha globin chains during red blood cell formation, while HBA2 acts as a backup and supportive gene to maintain stability.
How HBA1 and HBA2 Genes Work in Hemoglobin Production
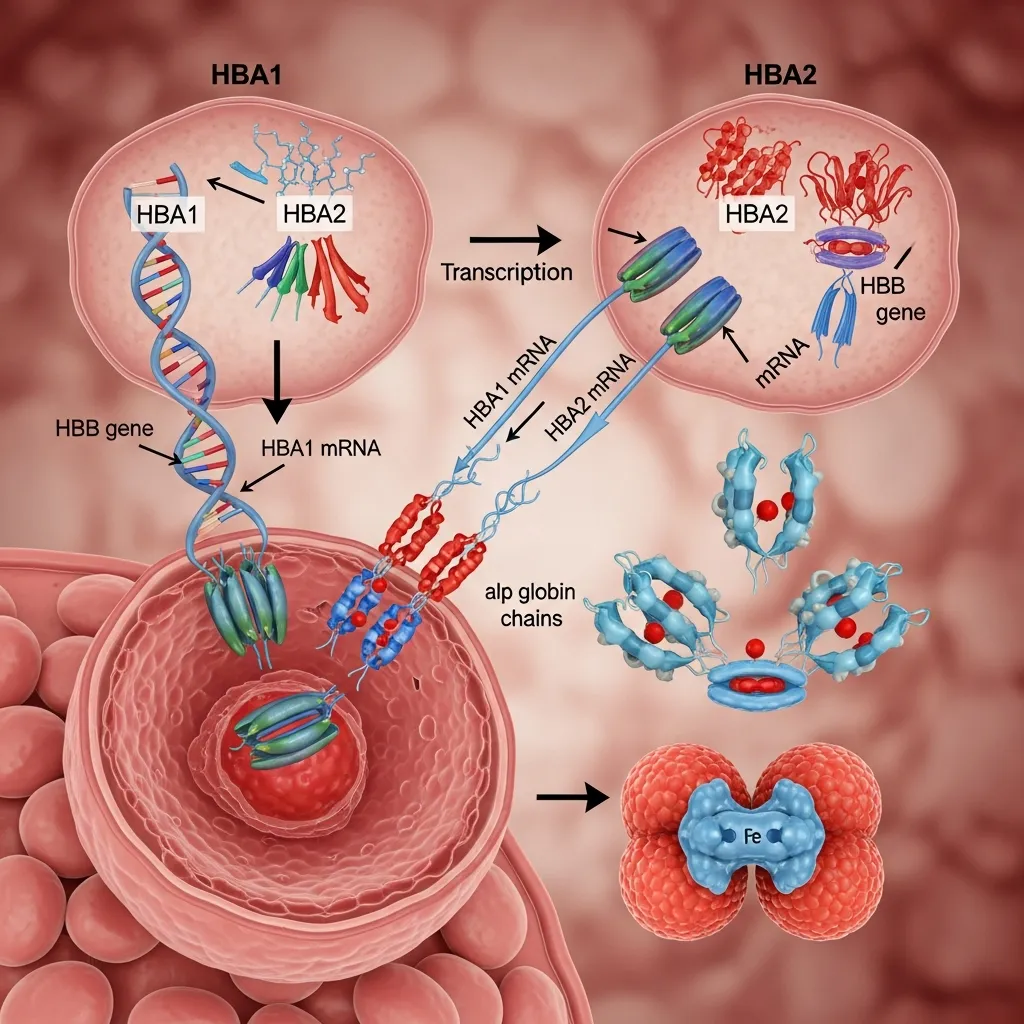 The HBA1 and HBA2 gene function becomes active during red blood cell formation in the bone marrow. These genes instruct cells to produce alpha globin chains, which combine with beta globin chains to form hemoglobin A (HbA), the most common type of hemoglobin in adults.
The HBA1 and HBA2 gene function becomes active during red blood cell formation in the bone marrow. These genes instruct cells to produce alpha globin chains, which combine with beta globin chains to form hemoglobin A (HbA), the most common type of hemoglobin in adults.
Without proper functioning of these genes, the balance between alpha and beta chains is disrupted, leading to unstable hemoglobin formation.
Key process includes:
- Gene activation in bone marrow
- Production of alpha globin chains
- Combination with beta globin chains
- Formation of functional hemoglobin
- Oxygen transport through the bloodstream
The alpha globin gene’s function in hemoglobin synthesis is essential for maintaining this balance and ensuring efficient oxygen delivery.
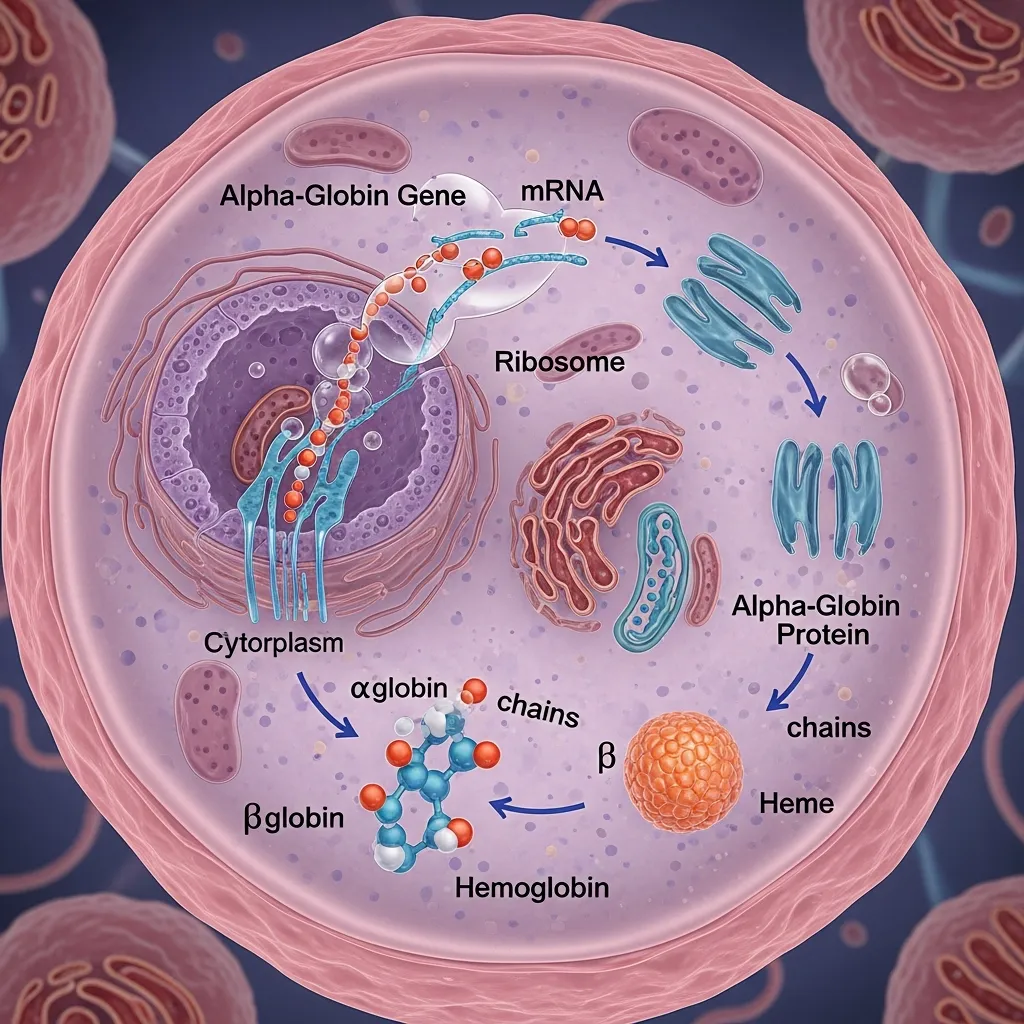 The alpha globin gene function in hemoglobin synthesis is to ensure that alpha chains are produced in equal amounts to beta chains. This balance is essential for forming stable hemoglobin molecules.
The alpha globin gene function in hemoglobin synthesis is to ensure that alpha chains are produced in equal amounts to beta chains. This balance is essential for forming stable hemoglobin molecules.If alpha globin production is reduced:
- Excess beta chains accumulate
- Abnormal hemoglobin forms
- Red blood cells become unstable
- Oxygen delivery decreases
This imbalance is a key cause of alpha thalassemia and related genetic blood disorders.
 The HBA1 and HBA2 gene function is extremely important in clinical genetics because it directly influences hemoglobin stability and overall red blood cell health. Any disruption in these genes can result in long-term or lifelong blood disorders that require careful monitoring, ongoing management, and in some cases, specialized treatment. Since these genes are responsible for producing alpha globin chains, even small genetic changes can significantly affect the body’s ability to maintain normal hemoglobin levels and oxygen transport efficiency.
The HBA1 and HBA2 gene function is extremely important in clinical genetics because it directly influences hemoglobin stability and overall red blood cell health. Any disruption in these genes can result in long-term or lifelong blood disorders that require careful monitoring, ongoing management, and in some cases, specialized treatment. Since these genes are responsible for producing alpha globin chains, even small genetic changes can significantly affect the body’s ability to maintain normal hemoglobin levels and oxygen transport efficiency.Conclusion
The HBA1 and HBA2 gene function is essential for producing alpha globin chains that form hemoglobin, the oxygen-carrying protein in blood. Proper functioning of these genes ensures healthy red blood cells and efficient oxygen delivery throughout the body.
Mutations or deletions in these genes can lead to serious conditions such as alpha thalassemia and hemoglobin disorders. Therefore, understanding their role is vital for early diagnosis, prevention, and genetic counseling.








{kind=link}