

")
The alpha globin gene deletion mechanism removes HBA1 or HBA2 genes through non-allelic homologous recombination during meiosis, causing alpha thalassemia.
Alpha thalassemia is one of the most common inherited blood disorders in the world, and most cases trace back to a single root cause: missing genetic material. The alpha globin gene deletion mechanism explains how the body loses the genes it needs to build healthy hemoglobin, and why some people develop severe anemia while others never notice a single symptom.
This guide breaks down exactly how these deletions happen at the DNA level. You will learn how the HBA1 and HBA2 genes work, why chromosome 16 is so prone to genetic errors, and how the number of deleted genes directly shapes the severity of alpha thalassemia. We will also cover diagnosis, treatment, and the questions families ask most often.
Whether you are a student, a healthcare professional, or someone with a family history of anemia, understanding the alpha globin gene deletion mechanism gives you the foundation to make informed decisions about screening, genetic counseling, and care.
What are alpha-globin genes and why do they matter?
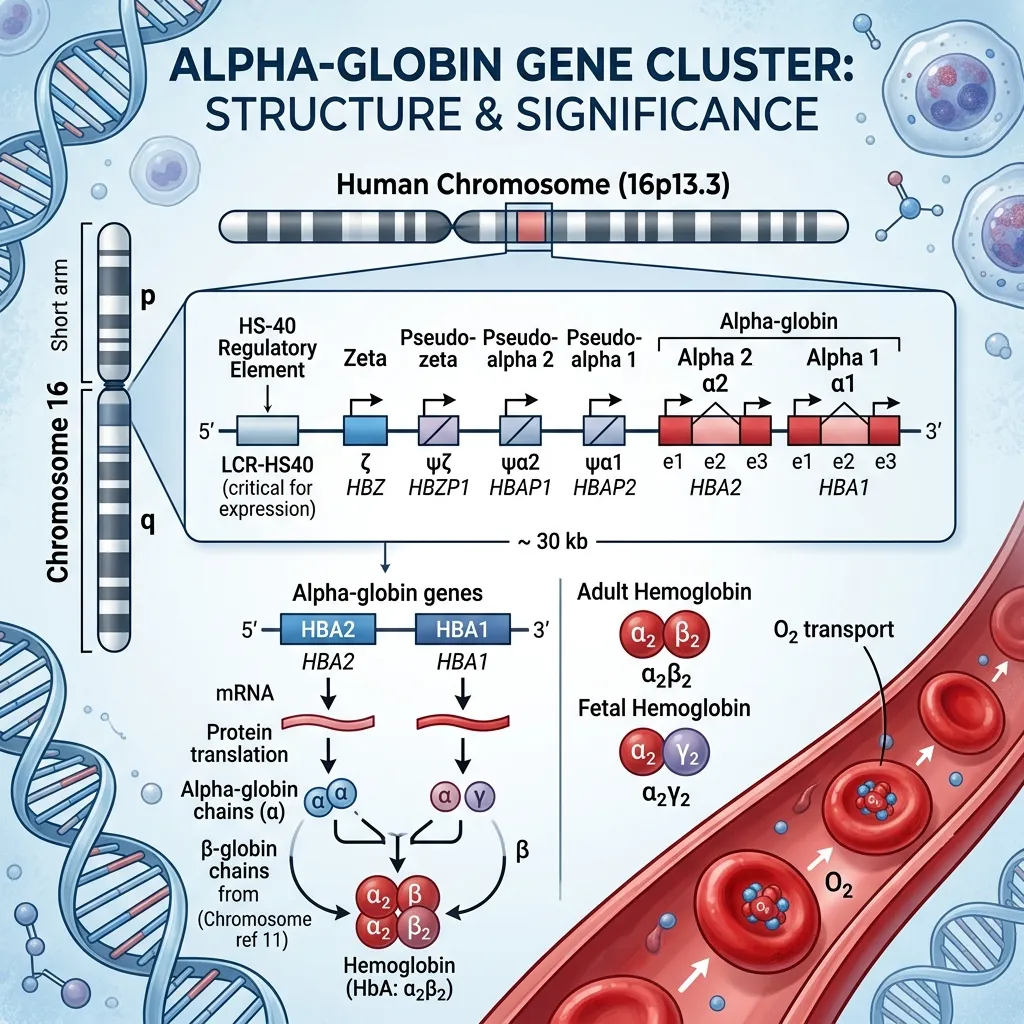 Alpha-globin genes carry the instructions your body uses to build alpha-globin chains, which are essential building blocks of hemoglobin. Hemoglobin is the protein inside red blood cells that carries oxygen from your lungs to every tissue and organ.
Alpha-globin genes carry the instructions your body uses to build alpha-globin chains, which are essential building blocks of hemoglobin. Hemoglobin is the protein inside red blood cells that carries oxygen from your lungs to every tissue and organ.
Healthy adult hemoglobin (HbA) is made of two alpha chains and two beta chains. This balanced structure keeps red blood cells stable and oxygen flowing efficiently. When alpha-globin production drops, the balance breaks down—and that imbalance sits at the heart of alpha thalassemia.
Thalassemia is not a single disease. It is a group of inherited blood disorders divided into two main types: alpha thalassemia, which affects alpha-globin production, and beta thalassemia, which affects beta-globin production. Alpha thalassemia is driven mostly by gene deletions, making it different from many other genetic conditions caused by small single-letter mutations.
Understanding the alpha globin gene deletion mechanism matters because it directly influences diagnosis, family planning, and treatment. For a deeper look at the genetics, our guide on the molecular basis of alpha thalassemia explores how these genetic changes disrupt hemoglobin at the cellular level.
How is the alpha-globin gene cluster organized?
The alpha-globin genes sit on the short arm of chromosome 16, near the telomere at position 16p13.3. This region is highly active during genetic recombination, which makes it a hotspot for deletions.
Where are HBA1 and HBA2 located?
Humans normally inherit four functional alpha-globin genes—two from each parent. These genes are called HBA1 and HBA2, and they sit very close together on chromosome 16. HBA1 and HBA2 share nearly identical DNA sequences and produce identical alpha-globin protein chains.
This high similarity is helpful for steady alpha-globin production, but it also creates a problem. Because the two genes look so alike, the chromosomes can misalign during cell division, setting the stage for deletion. Our detailed guide on HBA1 and HBA2 gene function explains how each gene contributes to hemoglobin production.
How does gene dosage affect hemoglobin synthesis?
A healthy person needs all four alpha-globin genes working together to produce enough alpha chains to match beta-chain production. This is called gene dosage. The more genes that are deleted, the less alpha-globin the body produces—and the more severe the resulting anemia becomes. This dose-dependent relationship is the key to understanding why alpha thalassemia ranges from completely silent to fatal before birth.
What is the alpha globin gene deletion mechanism?
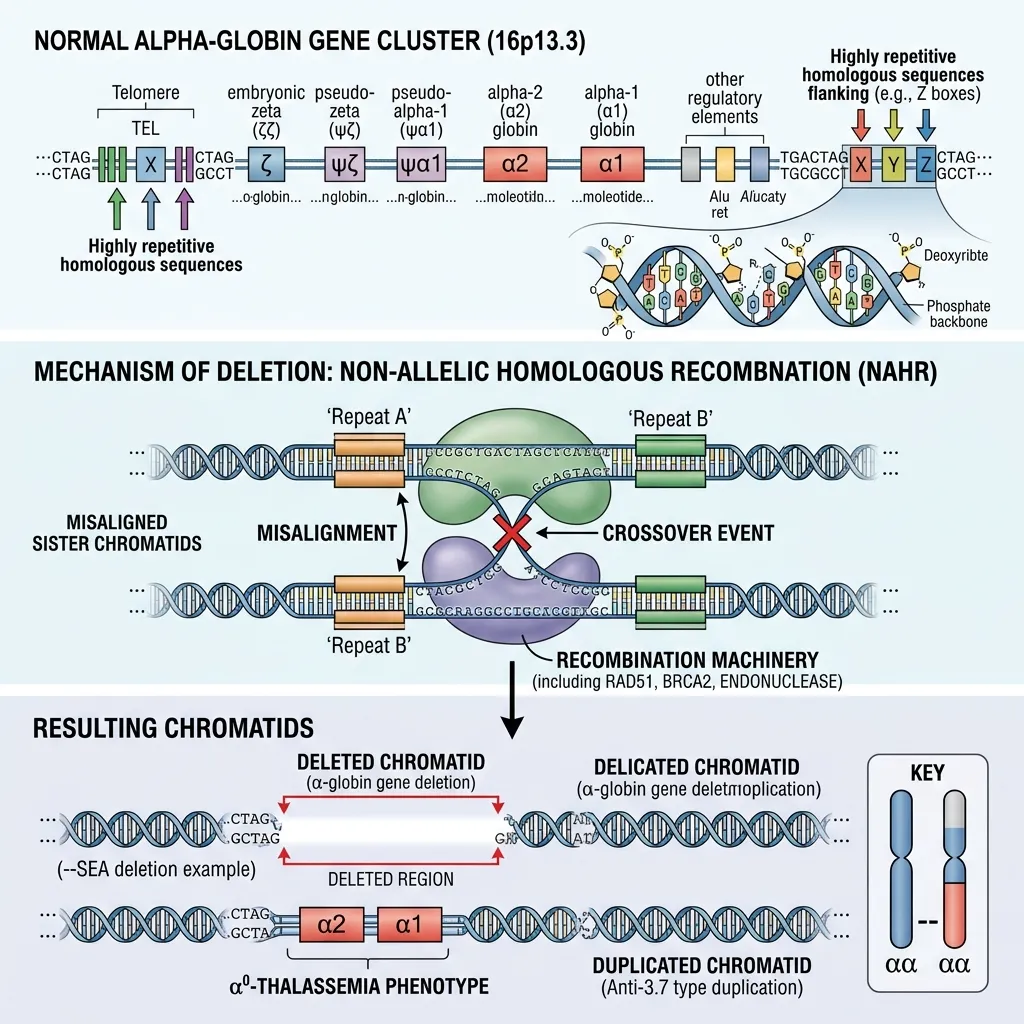 The alpha globin gene deletion mechanism primarily occurs through non-allelic homologous recombination (NAHR) during meiosis, the cell division that creates sperm and egg cells. This unequal crossing over results in the loss of HBA1 or HBA2 genes on chromosome 16.
The alpha globin gene deletion mechanism primarily occurs through non-allelic homologous recombination (NAHR) during meiosis, the cell division that creates sperm and egg cells. This unequal crossing over results in the loss of HBA1 or HBA2 genes on chromosome 16.
How does non-allelic homologous recombination cause deletions?
During meiosis, homologous chromosomes line up to swap genetic material. The alpha-globin gene cluster contains repetitive DNA sequences known as Z boxes and X boxes. Because these repeats look so similar, the chromosomes can misalign during pairing.
When misaligned chromosomes exchange genetic material, NAHR occurs. One chromosome ends up with extra genetic material (a duplication), while the other loses material (a deletion). The chromosome carrying the deletion passes the alpha thalassemia gene mutation on to the next generation. This is the most frequent driver of alpha thalassemia worldwide.
What are the most common deletion types?
Doctors classify alpha-globin deletions by the specific DNA segments that are lost:
- Single-gene deletions (-α3.7 and -α4.2): These remove one alpha gene and leave one functional gene on the affected chromosome. They are the most common deletions seen worldwide.
- Double-gene deletions (–SEA and –MED): The Southeast Asian (–SEA) and Mediterranean (–MED) deletions remove both HBA1 and HBA2 from a single chromosome, leading to more serious clinical outcomes.
What less common deletion mechanisms exist?
While unequal crossing over explains most cases, the mechanism of alpha thalassemia gene mutation can also involve rarer events. Larger gross deletions can remove regulatory regions that control alpha-globin genes, even when the genes themselves remain intact. Repetitive DNA sequences across chromosome 16 make these structural errors more likely, which is why this region is considered a recombination hotspot.
How does alpha globin gene deletion cause thalassemia?
Alpha globin gene deletion causes thalassemia by reducing the supply of alpha-globin chains needed to build stable hemoglobin. When alpha chains run short, beta chains (in adults) or gamma chains (in fetuses) are left unpaired. These unpaired chains form unstable clusters that damage red blood cell membranes, leading to premature cell destruction and chronic anemia.
The number of deleted genes directly determines severity. This clear genotype-to-phenotype relationship is one of the most predictable patterns in genetic medicine.
Single gene deletion: silent carrier
Losing one of four genes (-α/αα) produces a silent carrier. These individuals have normal hemoglobin levels and no symptoms. The condition is usually found only through genetic screening.
Two gene deletion: alpha thalassemia trait
Losing two genes causes alpha thalassemia trait, also called alpha thalassemia minor. Patients typically have mild microcytic anemia with small, pale red blood cells. Many are misdiagnosed with iron deficiency. Our complete guide on alpha thalassemia trait explains testing and care in detail.
Three gene deletion: hemoglobin H disease
Losing three genes (–/-α) causes hemoglobin H disease. The severe shortage of alpha chains lets excess beta chains form Hemoglobin H. Patients face moderate to severe hemolytic anemia, an enlarged spleen, and sometimes bone changes, requiring ongoing medical care.
Four gene deletion: hydrops fetalis
Losing all four genes (–/–) is the most severe form. With no alpha-globin at all, the fetus produces Hemoglobin Barts, leading to massive fluid buildup and heart failure. This condition, called hydrops fetalis, is usually fatal before or shortly after birth.
What are the HBA1 HBA2 gene deletion effects in alpha thalassemia?
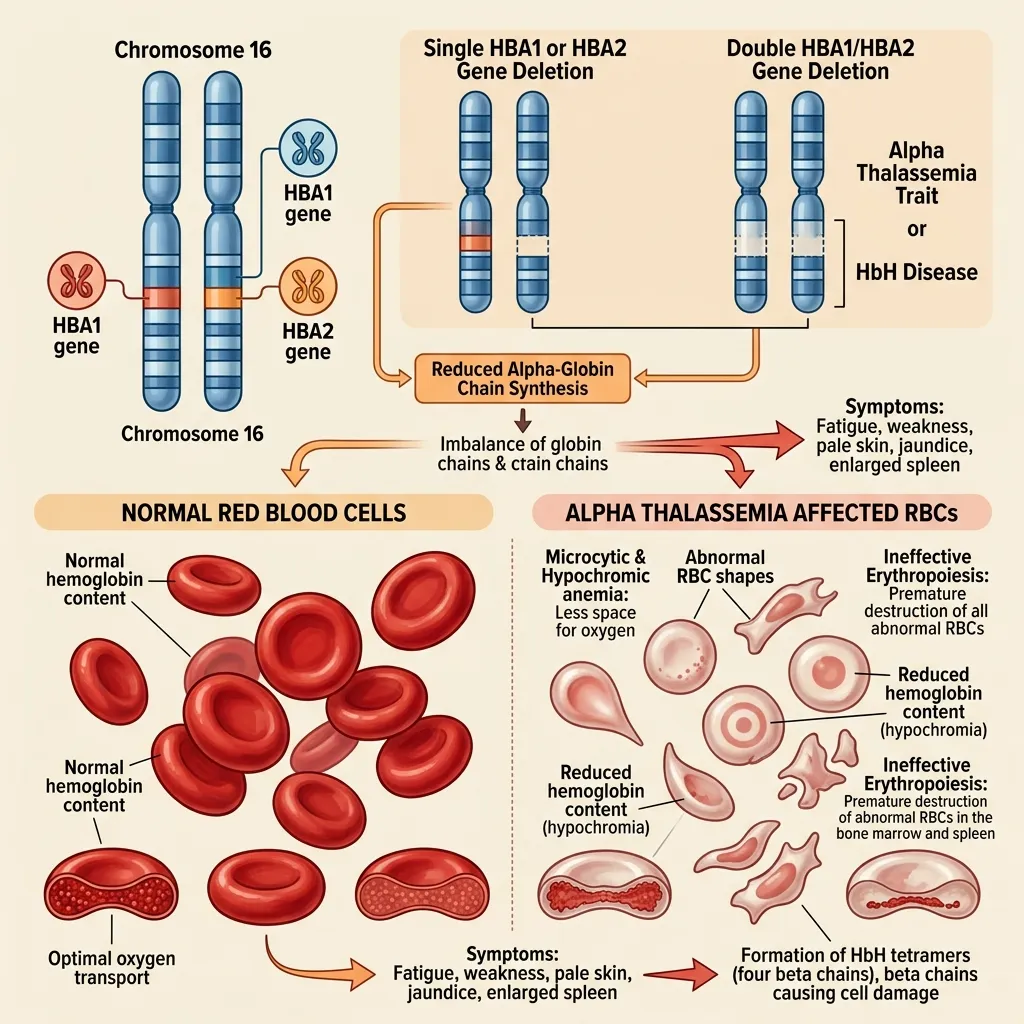 The HBA1 HBA2 gene deletion effects in alpha thalassemia depend on which genes are lost and how many. Because HBA1 and HBA2 produce identical proteins, the total count of working genes matters more than which specific gene is deleted.
The HBA1 HBA2 gene deletion effects in alpha thalassemia depend on which genes are lost and how many. Because HBA1 and HBA2 produce identical proteins, the total count of working genes matters more than which specific gene is deleted.
When deletions remove these genes, the body produces fewer alpha chains, the alpha-to-beta ratio collapses, and unstable hemoglobin forms. This drives the symptoms that define the condition: chronic anemia, fatigue, weakness, increased cardiac workload, and—in severe cases—organ stress from long-term low oxygen. The specific combination of deletions a person inherits dictates exactly where they fall on this clinical spectrum.
How do non-deletion mutations compare?
Not all alpha thalassemia comes from missing genes. A smaller share of cases stems from non-deletion mutations, where the gene stays physically present but stops working properly.
These include point mutations (single base-pair changes), splice site mutations, and mutations in regulatory regions that control how genes turn on. Unlike full deletions, these mutations often allow partial alpha-globin production. As a result, disease severity varies widely depending on the mutation type.
The key difference: deletions remove genetic material entirely, while non-deletion mutations disrupt how existing genes function. Both lead to alpha-globin imbalance, but non-deletion forms can sometimes produce more variable clinical pictures. Together, deletions and point mutations form the full genetic foundation of alpha thalassemia.
Treatment depends entirely on severity, which the deletion count predicts.
- Silent carriers and trait: No treatment needed. Importantly, these patients should avoid unnecessary iron supplements, since their anemia is genetic, not iron-related.
- Hemoglobin H disease: Care often includes folic acid supplementation, periodic blood transfusions during anemic crises, and sometimes splenectomy.
- Hydrops fetalis: Historically fatal, this condition can occasionally be managed with intrauterine transfusions, followed by lifelong transfusion therapy if the infant survives.
Genetic counseling is vital for at-risk families. Our family planning thalassemia guide covers carrier screening, prenatal diagnosis, and reproductive options like IVF with preimplantation genetic diagnosis.
Looking ahead, gene therapy offers the most promising future. Researchers are developing techniques to insert functional alpha-globin genes into a patient’s own stem cells—a potential path to curing the condition without lifelong transfusions.
Why understanding this mechanism matters
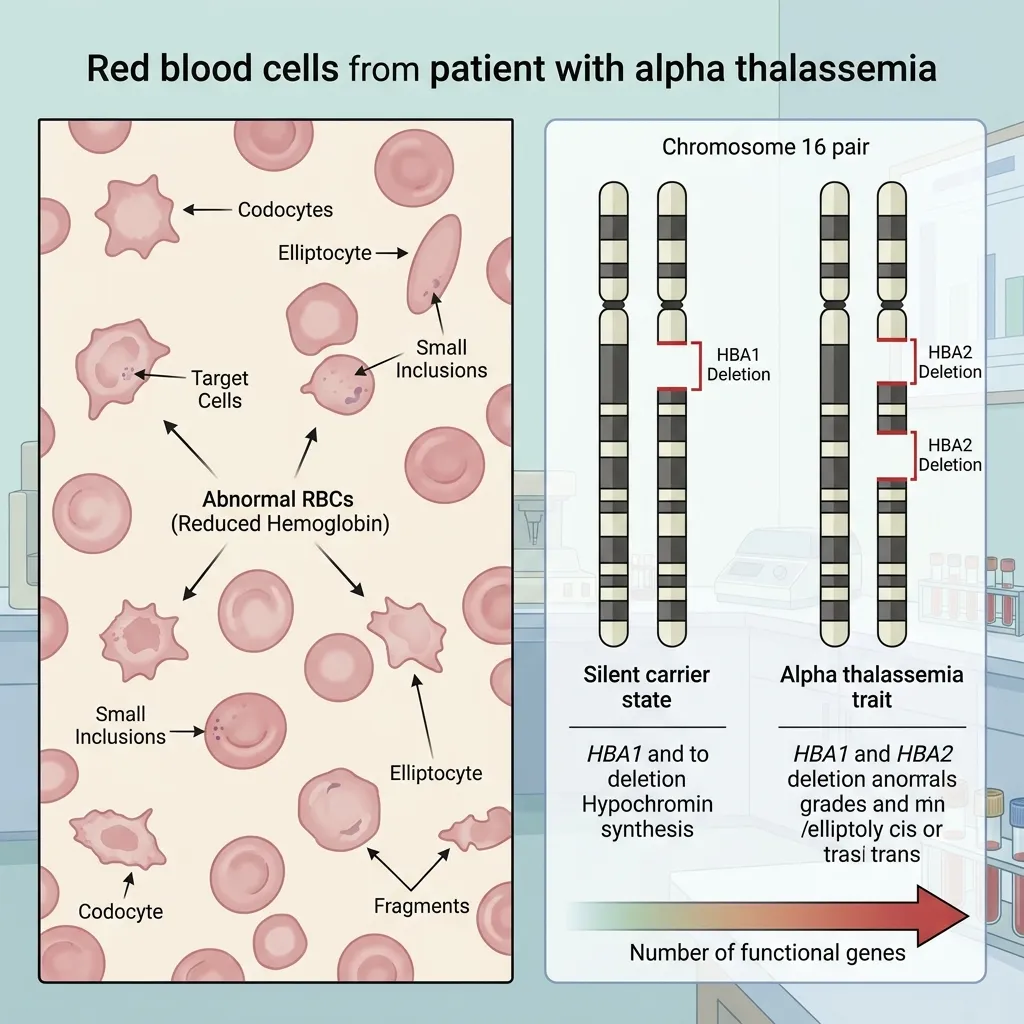 The alpha globin gene deletion mechanism is a complex but highly predictable process, driven by the unique structure of chromosome 16. Unequal crossing over and non-allelic homologous recombination strip away the HBA1 and HBA2 genes, producing a spectrum that runs from silent carriers to fatal fetal conditions.
The alpha globin gene deletion mechanism is a complex but highly predictable process, driven by the unique structure of chromosome 16. Unequal crossing over and non-allelic homologous recombination strip away the HBA1 and HBA2 genes, producing a spectrum that runs from silent carriers to fatal fetal conditions.
Grasping how alpha globin gene deletion causes thalassemia helps healthcare providers deliver accurate diagnoses, precise counseling, and timely care. As gene therapy and advanced molecular diagnostics evolve, the medical community moves closer to reducing the severe burden of this disorder.
If you or your partner has a family history of anemia, the most valuable next step is simple: consult a hematologist or genetic counselor about comprehensive genetic screening. Early knowledge gives families the widest range of choices and the most time to plan. For authoritative global guidance, the World Health Organization and the Centers for Disease Control and Prevention offer trusted resources on thalassemia screening and care.








{kind=link}