


Hemoglobin Barts is a severe form of alpha-thalassemia caused by the complete absence of alpha-globin genes, leading to extreme oxygen delivery failure in the body. It is often detected before birth and can cause serious complications such as hydrops fetalis. While historically fatal, advances in prenatal care, transfusions, and gene therapy for thalassemia are improving outcomes and offering new hope for future treatment.
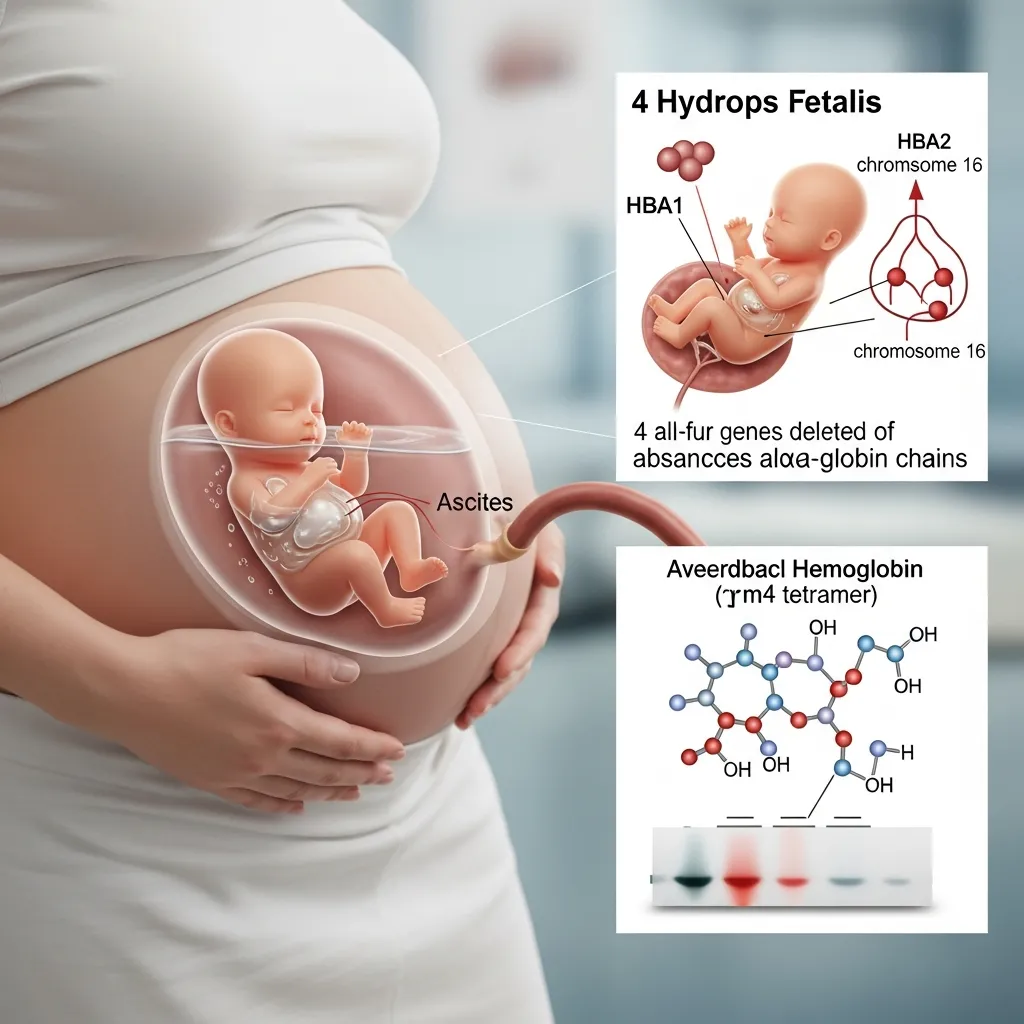 To understand Hemoglobin Barts, we first need to look at the genetic instructions that build normal hemoglobin. Healthy adult hemoglobin consists of two alpha-globin proteins and two beta-globin proteins. The instructions for making alpha-globin are located on chromosome 16. Every person inherits four alpha-globin genes—two from their mother and two from their father.
To understand Hemoglobin Barts, we first need to look at the genetic instructions that build normal hemoglobin. Healthy adult hemoglobin consists of two alpha-globin proteins and two beta-globin proteins. The instructions for making alpha-globin are located on chromosome 16. Every person inherits four alpha-globin genes—two from their mother and two from their father.Alpha-thalassemia occurs when one or more of these four alpha-globin genes are missing or mutated. The severity of the disorder depends entirely on how many genes are affected. If a person is missing one or two genes, they might only experience mild alpha thalassemia trait symptoms, such as slight fatigue or mild anemia. They usually live normal, healthy lives without needing medical treatment.
However, when a developing fetus inherits mutations causing the loss of all four alpha-globin genes, the body cannot produce any alpha-globin proteins. Without alpha-globin, the remaining gamma-globin proteins (which are present during fetal development) clump together in groups of four. This abnormal cluster of four gamma-globin proteins is called Hemoglobin Barts.
Because Hemoglobin Barts has an extremely high affinity for oxygen, it holds onto the oxygen tightly and refuses to release it to the developing tissues. This leads to a severe, life-threatening form of alpha-thalassemia known as Hemoglobin Barts Hydrops Fetalis Syndrome. The extreme oxygen starvation causes severe anemia, heart failure, and a massive buildup of fluid in the fetus’s body.
If the baby is born prematurely or without a prior prenatal diagnosis, postnatal confirmation involves taking a blood sample immediately. A test called hemoglobin electrophoresis separates the different types of hemoglobin in the blood, clearly identifying the high levels of Hemoglobin Barts.
Management and Treatment Options for Infants
 Historically, Hemoglobin Barts Hydrops Fetalis was considered a uniformly fatal condition, with most infants passing away before or shortly after birth. Today, rapid advancements in maternal-fetal medicine have opened up new possibilities for treatment, though the journey remains highly complex and demanding.
Historically, Hemoglobin Barts Hydrops Fetalis was considered a uniformly fatal condition, with most infants passing away before or shortly after birth. Today, rapid advancements in maternal-fetal medicine have opened up new possibilities for treatment, though the journey remains highly complex and demanding.
Intrauterine Interventions
When Hemoglobin Barts is diagnosed early in the pregnancy, doctors may offer intrauterine blood transfusions. This highly specialized procedure involves injecting healthy, oxygen-rich red blood cells directly into the umbilical cord vein of the fetus. These transfusions can correct the severe fetal anemia, reduce the fluid buildup associated with hydrops, and allow the pregnancy to continue closer to full term.
This intervention carries significant risks, including premature labor or infection, and requires a highly skilled medical team. However, successful intrauterine transfusions have allowed many infants with Hemoglobin Barts to survive to birth.
Postnatal Care and Lifelong Transfusions
Infants born with Hemoglobin Barts who survive delivery require immediate, intensive neonatal care. They are often born prematurely and may need mechanical ventilation to help them breathe.
Because their bodies still cannot produce normal hemoglobin, these children require lifelong, regular blood transfusions every three to four weeks. These transfusions supply the healthy red blood cells needed to deliver oxygen to their growing bodies.
Frequent blood transfusions introduce a new complication: iron overload. The body cannot naturally excrete the excess iron contained in the transfused blood. Over time, toxic levels of iron build up in vital organs like the heart, liver, and pancreas. To combat this, patients must undergo daily iron chelation therapy, taking medications that bind to the excess iron and help flush it out of the system.
Stem Cell Transplantation
Currently, the only curative treatment for Hemoglobin Barts is a hematopoietic stem cell transplant (bone marrow transplant). This procedure replaces the patient’s defective blood-forming stem cells with healthy stem cells from a matched donor, often a sibling. If successful, the new stem cells will begin producing healthy red blood cells with normal hemoglobin, eliminating the need for lifelong transfusions.
Long-Term Prognosis and Quality of Life
The long-term prognosis for individuals surviving Hemoglobin Barts has improved significantly over the last few decades, thanks to aggressive early intervention and better management of iron overload. However, living with a severe chronic blood disorder requires immense resilience and strict adherence to medical protocols.
Children who undergo successful stem cell transplants can go on to live normal, healthy lives without the burden of chronic anemia. For those who rely on lifelong transfusions, the quality of life depends heavily on access to high-quality medical care and effective iron chelation therapy.
Without proper management, severe anemia causes the bone marrow to expand dangerously as it attempts to produce more red blood cells. This bone marrow hyperplasia can lead to skeletal deformities, such as chipmunk facies, where the facial bones become prominent and the jaw misaligns. Regular transfusions prevent the bone marrow from overworking, thereby protecting the patient’s bone structure and supporting normal physical growth.
Psychological support is equally important. Managing a demanding medical schedule can take a toll on patients and their families. Connecting with support groups, engaging in counseling, and leaning on a multidisciplinary healthcare team helps families navigate the emotional challenges of chronic illness.
Research Advancements and Future Directions

The scientific community is actively exploring innovative ways to treat severe hemoglobinopathies, offering hope for safer and more accessible cures in the near future.
One of the most promising areas of research is gene therapy. Unlike a traditional stem cell transplant, which requires a matched donor, gene therapy uses the patient’s own stem cells. Scientists extract these cells, use advanced technology to insert functional copies of the missing alpha-globin genes, and then infuse the corrected cells back into the patient. Early clinical trials for various forms of thalassemia are showing encouraging results, pointing toward a future where patients can be cured using their own biology.
Global health organizations, such as the World Health Organization (WHO), continue to advocate for better screening programs and standardized care protocols across different countries. Increased funding for research is vital. This is where community action plays a massive role. By engaging in digital advocacy for thalassemia, individuals can raise public awareness, promote blood donation, and support policies that fund groundbreaking genetic research.
Empowering Families Through Early Detection and Care
Receiving a diagnosis of Hemoglobin Barts is a life-altering event. The condition presents profound medical challenges, from the complexities of intrauterine transfusions to the daily management of a chronic illness. Yet, the narrative surrounding this severe form of alpha-thalassemia is slowly changing.
Advancements in prenatal screening allow families to prepare and explore early interventions. Improved transfusion protocols and iron chelation therapies are extending life expectancies and enhancing the quality of life for survivors. With the horizon of gene therapy drawing closer, there is genuine hope for a universal cure.
If you or your partner have a family history of blood disorders, seek out genetic counseling before or early in your pregnancy. Knowledge is your most powerful tool. By understanding the genetic risks and working closely with specialized maternal-fetal medical teams, families can navigate the complexities of Hemoglobin Barts with proactive care and comprehensive support.








{kind=link}