


Molecular basis of alpha thalassemia explains genetic mutations, gene deletions, and hemoglobin defects affecting red blood cell production and oxygen transport.
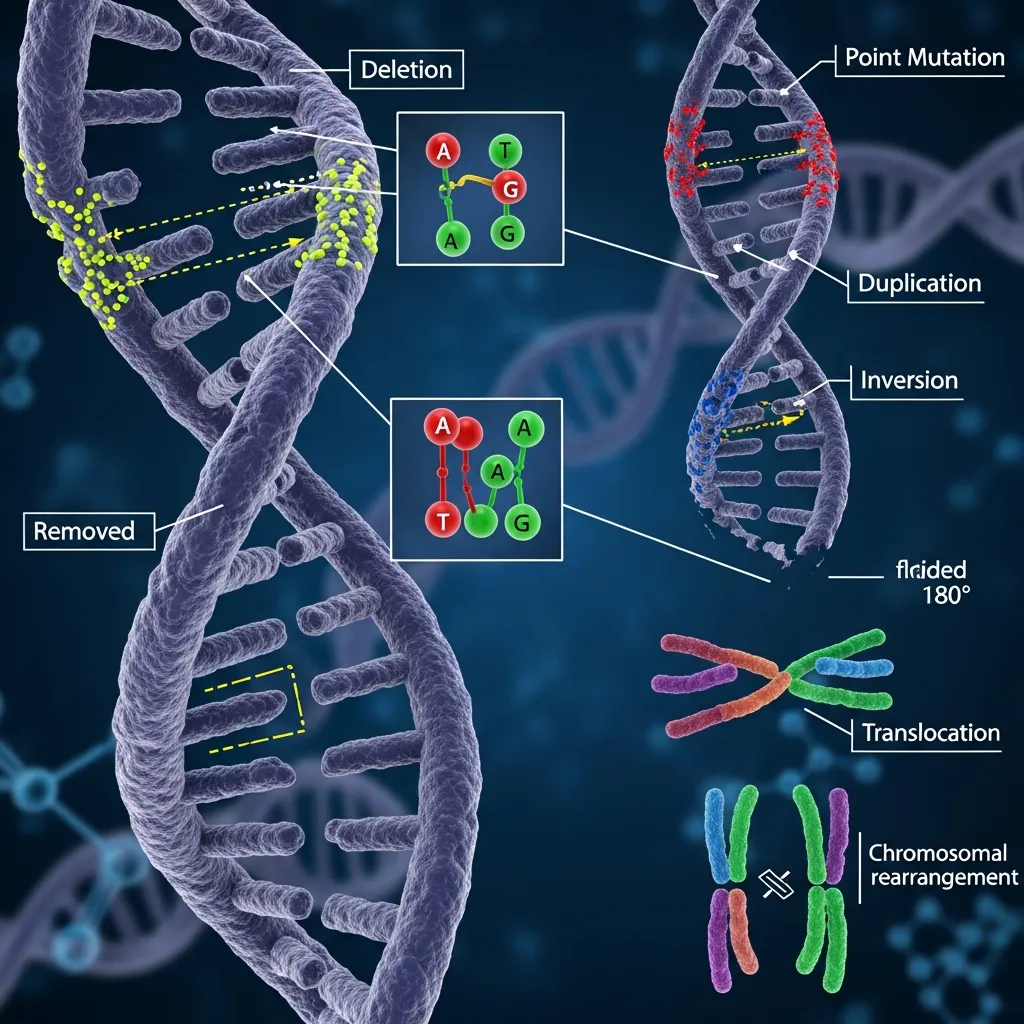 The molecular defects in alpha thalassemia can be broadly classified into two main categories:
The molecular defects in alpha thalassemia can be broadly classified into two main categories:
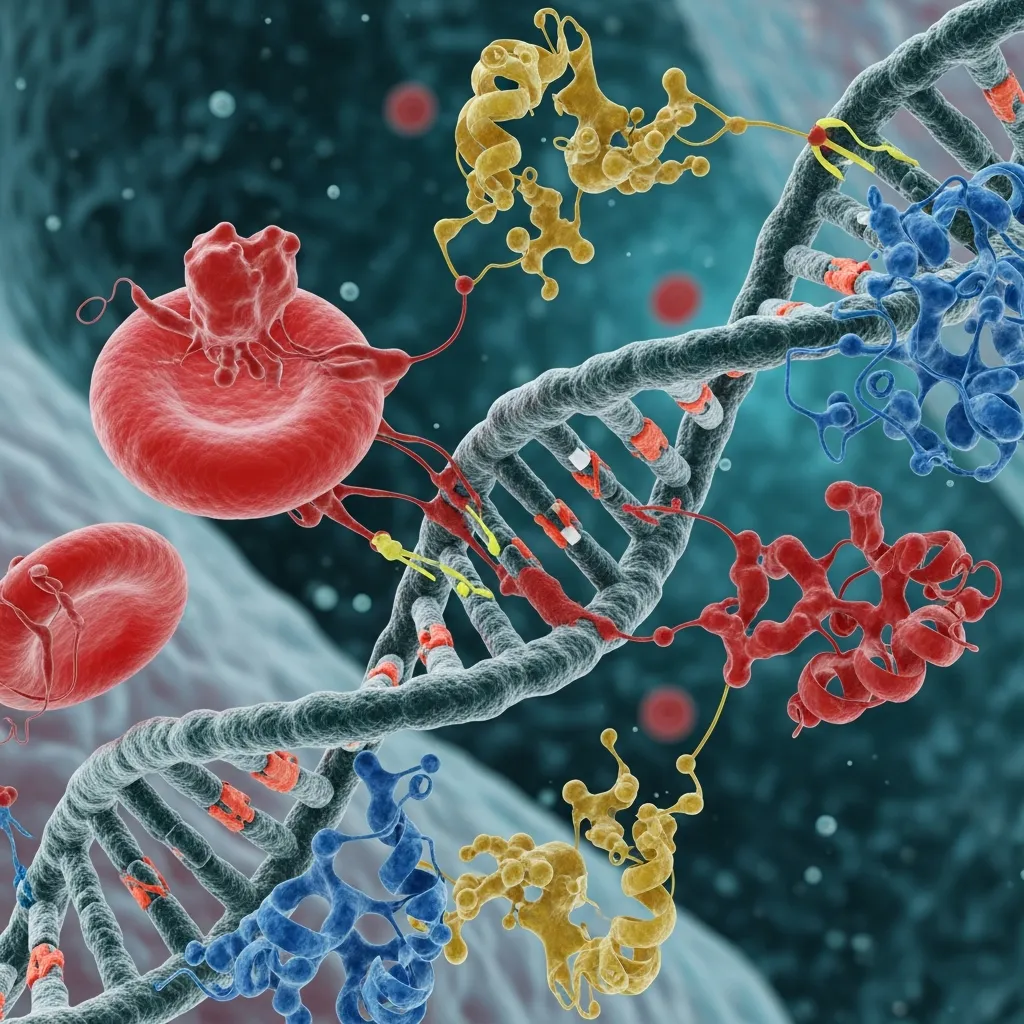 Apart from deletions, point mutations also contribute to the Molecular basis of alpha thalassemia. These mutations may not completely remove the gene but instead affect how well it functions.
Apart from deletions, point mutations also contribute to the Molecular basis of alpha thalassemia. These mutations may not completely remove the gene but instead affect how well it functions.
Alpha Thalassemia Molecular Pathophysiology
The Alpha thalassemia molecular pathophysiology explains how genetic defects at the DNA level translate into clinical symptoms observed in patients. It describes the step-by-step biological process that connects gene abnormalities with abnormal hemoglobin production and red blood cell destruction.
Step 1: Reduced Alpha Globin Production
The process begins with gene deletions or mutations affecting HBA1 or HBA2 genes. This results in decreased or absent alpha globin chain synthesis, which is essential for normal hemoglobin formation.
Step 2: Chain Imbalance
As alpha chains decrease, excess beta chains accumulate in adults. This imbalance disrupts the normal alpha-to-beta ratio required for stable hemoglobin (HbA).
Step 3: Abnormal Hemoglobin Formation
In more severe cases, excess beta chains form unstable tetramers known as HbH (β4). These abnormal molecules cannot effectively carry oxygen and are highly unstable.
Step 4: Red Blood Cell Damage
Unstable hemoglobin damages red blood cell (RBC) membranes, making them fragile. This leads to structural deformities and reduced cell lifespan.
Step 5: Hemolysis
Damaged RBCs undergo premature destruction (hemolysis), leading to chronic anemia. This hemolytic process is a central feature of the Molecular basis of alpha thalassemia.
Hemoglobin Imbalance and Its Effects
When alpha chains are missing, the body cannot form stable hemoglobin.
This leads to multiple physiological consequences:
- Reduced oxygen delivery to tissues
- Chronic anemia
- Fatigue and generalized weakness
- Increased cardiac workload
- Organ stress due to long-term hypoxia
In severe cases, especially when all alpha genes are affected, fetal oxygen transport fails, leading to life-threatening complications such as hydrops fetalis.
Clinical Spectrum of Alpha Thalassemia
The severity of alpha thalassemia depends on how many alpha globin genes are affected:
Silent Carrier (1 gene missing)
- No symptoms
- Normal life expectancy
- Often undetected without genetic testing
Alpha Thalassemia Trait (2 genes missing)
- Mild anemia
- Microcytosis
- Frequently undiagnosed or mistaken for iron deficiency
👉 Read more:
https://thalassemiaawarenet.link/alpha-thalassemia-trait-complete-guide/
HbH Disease (3 genes missing)
- Moderate to severe anemia
- Enlarged spleen may occur
- Requires regular medical management
Hydrops Fetalis (4 genes missing)
- Severe fetal anemia
- Usually fatal before or shortly after birth
- Medical emergency in pregnancy
 The Molecular basis of alpha thalassemia follows an autosomal recessive inheritance pattern, which explains how the condition is passed from parents to children. This genetic pattern is central to understanding why the disease appears in different severity levels within families.
The Molecular basis of alpha thalassemia follows an autosomal recessive inheritance pattern, which explains how the condition is passed from parents to children. This genetic pattern is central to understanding why the disease appears in different severity levels within families. Alpha thalassemia is one of the most common inherited blood disorders worldwide, with particularly high prevalence in regions where malaria has historically been endemic. These areas include Southeast Asia, the Mediterranean basin, the Middle East, and parts of Africa. In these populations, the carrier frequency can be very high, making alpha thalassemia a significant public health concern.
Alpha thalassemia is one of the most common inherited blood disorders worldwide, with particularly high prevalence in regions where malaria has historically been endemic. These areas include Southeast Asia, the Mediterranean basin, the Middle East, and parts of Africa. In these populations, the carrier frequency can be very high, making alpha thalassemia a significant public health concern.Conclusion
The Molecular basis of alpha thalassemia is rooted in genetic deletions and mutations that disrupt normal alpha globin production. This imbalance leads to abnormal hemoglobin formation, ineffective red blood cell production, and varying degrees of anemia.
Understanding Alpha globin gene deletion mechanisms, Genetic mutations in alpha thalassemia, and Alpha thalassemia molecular pathophysiology is essential for accurate diagnosis, prevention, and management.
Advances in genetic testing and molecular biology have made it possible to detect the disease early and guide family planning decisions effectively. With proper awareness and screening, severe forms of alpha thalassemia can be significantly reduced.








{kind=link}